
Abstract
The nuclear factor κB (NF-κB) pathway is critical for regulating immune and inflammatory responses, and uncontrolled NF-κB activation is closely associated with various inflammatory diseases and malignant tumors. The Met1-linked linear ubiquitin chain, which is generated by linear ubiquitin chain assembly complex (LUBAC), is important for regulating NF-κB activation. This process occurs through the linear ubiquitination of NF-κB essential modulator, a regulatory subunit of the canonical inhibitor of the NF-κB kinase complex. In this study, we have established a robust and efficient high-throughput screening (HTS) platform to explore LUBAC inhibitors, which may be used as tool compounds to elucidate the pathophysiological role of LUBAC. The HTS platform consisted of both cell-free and cell-based assays: (1) cell-free LUBAC-mediated linear ubiquitination assay using homogenous time-resolved fluorescence technology and (2) cell-based LUBAC assay using the NF-κB luciferase reporter gene assay. By using the HTS platform, we performed a high-throughput chemical library screen and identified several hit compounds with selectivity against a counterassay. Liquid chromatography–mass spectrometry analysis revealed that these compounds contain a chemically reactive lactone structure, which is transformed to give reactive α,β-unsaturated carbonyl compounds. Further investigation revealed that the reactive group of these compounds is essential for the inhibition of LUBAC activity.
Introduction
The ubiquitin system functions as a pivotal posttranscriptional modification system in numerous cellular functions, including protein degradation, membrane trafficking, DNA repair, and signal transduction.1,2 Ubiquitination is catalyzed by three kinds of enzymes: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3). 3 In this process, the C-terminus of ubiquitin is first activated by E1, using the high energy produced by hydrolysis of ATP to AMP, and then is conjugated to a cysteine residue in the active site of E1 through a reactive thioester bond. The ubiquitin is subsequently transferred to an active cysteine residue of E2. E3 selectively recognizes both substrates and E2 and catalyzes the transfer of ubiquitin from E2 primarily to the ε-amino group of lysine residues in target proteins via an isopeptide bond. Finally, deubiquitinases remove the ubiquitins from the target proteins, and the ubiquitins are recycled.
It is believed that polyubiquitin chains are formed only by the conjugation of the ubiquitin C-terminus glycine to an internal lysine residue of another ubiquitin through the formation of an isopeptide bond. However, it has recently been reported that a new type of linear polyubiquitin chain exists in which the C-terminus glycine of ubiquitin is conjugated to the α-amino group of the N-terminus methionine residue of another ubiquitin. This linear polyubiquitin is generated by a unique ubiquitin ligase complex named linear ubiquitin chain assembly complex (LUBAC). 4 LUBAC forms a 600 kDa physiological ternary complex composed of a longer isoform of heme-oxidized iron-regulatory protein ubiquitin ligase 1 (HOIL-1L), HOIL-1L interacting protein (HOIP), and Shank-associated RH domain-interacting protein (SHARPIN). It specifically regulates the canonical nuclear factor κB (NF-κB) pathway via the linear ubiquitination of NF-κB essential modulator (NEMO), a regulatory subunit of the canonical inhibitor of NF-κB kinase (IKK) complex, and receptor-interacting protein 1.
The NF-κB pathway is critical for the regulation of immune and inflammatory responses, and uncontrolled NF-κB activation is closely associated with various inflammatory diseases and malignant tumors. In the absence of LUBAC components, NF-κB signaling is attenuated and induces apoptosis and inflammation.5–8 Many studies on the pathophysiological functions of LUBAC, including B- and T-cell development, innate and adaptive immune response,9–14 carcinogenesis, 15 and osteogenesis 16 in humans, have recently been performed. However, the relevance and functions of LUBAC in pathogenesis remain controversial.
To better understand the mechanism of LUBAC-mediated NF-κB activation and its role under normal and pathophysiological conditions, compounds selectively activating or inhibiting LUBAC may be useful pharmacological tools. In particular, small-molecular-weight LUBAC inhibitors may be novel therapeutic agents for the treatment of diseases associated with NF-κB hyperactivation.
Previously, a time-resolved fluorescence resonance energy transfer (TR-FRET)-based assay was developed to detect LUBAC-mediated linear polyubiquitin chain elongation, and the fungal metabolite gliotoxin was identified as a selective LUBAC inhibitor. 17 However, owing to its cytotoxicity, the use of this compound was found to be limited. In this study, we developed a robust and efficient high-throughput screening (HTS) platform to screen for selective LUBAC inhibitors, which may be used as tool compounds to elucidate the pathophysiological role of LUBAC. The HTS platform consisted of both cell-free and cell-based assays: (1) a cell-free LUBAC-mediated linear ubiquitination assay using homogenous time-resolved fluorescence (HTRF) technology, along with a counterassay using an HTRF-based p-IκBα polyubiquitination assay, and (2) a cell-based NF-κB luciferase reporter gene assay in LUBAC-overexpressing cells, along with a counterassay using NEMO fused with three linear ubiquitin moieties at its C-terminus NEMO-(Ubi)3-overexpressing cells. By using the HTS platform, we were able to efficiently identify novel and selective LUBAC inhibitors.
Material and Methods
Compound Synthesis
JTP-1048196, 3-[2-(2-methoxyphenyl)-2-oxoethyl]-2-benzofuran-1(3H)-one, and JTP-0819958, sodium 2-[(1E)-3-(2-methoxyphenyl)-3-oxoprop-1-en-1-yl]benzoate, were synthesized as described in Supplemental Figure S1 .
Plasmids and Baculoviruses
DNA fragments encoding human HOIL-1L, HOIP, SHARPIN, ubiquitin-activating enzyme E1 (Ube1), RING-box protein 1 (Rbx1), NEMO, and ubiquitin (Ubi) were amplified from Human HeLa cells or Liver Spleen Marathon-Ready cDNA (Takara Bio, Shiga, Japan) by PCR with specific primer pairs. Human ubiquitin-conjugating enzyme H5c (UbcH5c) and human IκBα cDNA were purchased from Origene Technologies (Rockville, MD). They were subsequently subcloned into pcDNA3.1(+) (Thermo Fisher Scientific, Waltham, MA), pVL1393 (Becton, Dickinson and Company, Franklin Lakes, NJ), pET-17b (Merck KGaA, Darmstadt, Germany), pET-28a (Merck KGaA), or pGEX-6P-1 (GE Healthcare, Buckinghamshire, England) vectors, which resulted in the following constructs: pcDNA3.1(+)/HOIL-1L-Flag pcDNA3.1(+)/HOIL-1L-hemagglutinin (HA) pcDNA3.1(+)/HOIL-1L (ΔC; residues 1–191)-Flag pcDNA3.1(+)/His-HOIP pcDNA3.1(+)/HA-HOIP pcDNA3.1(+)/His-HOIP (ΔN; residues 474–1072) pcDNA3.1(+)/Flag-SHARPIN pcDNA3.1(+)/HA-SHARPIN pcDNA3.1(+)/HA-NEMO pcDNA3.1(+)/HA-NEMO-Ubi(G76V)-Ubi(G76V)-Ubi(G75,76A), referred to as pcDNA3.1(+)/HA-NEMO-(Ubi)3 pVL1393/HOIL-1L-Flag pVL1393/HOIL-1L (ΔC)-Flag pVL1393/His-HOIP pVL1393/His-HOIP (ΔN) pVL1393/Flag-SHARPIN pVL1393/His-Ube1 pVL1393/His-Rbx1 pET-17b/Ubi-glutathione S-transferase (GST) pET-28a/UbcH5c pGEX-6P-1/IκBα
Recombinant baculoviruses encoding HOIL-1L-Flag, HOIL-1L (ΔC)-Flag, His-HOIP, His-HOIP (ΔN), Flag-SHARPIN, His-Ube1, and His-Rbx1 were generated using the BD BaculoGold baculovirus expression system (Becton, Dickinson and Company). Recombinant baculoviruses encoding His-tagged β-transducin repeat-containing proteins (His-TrCP1), Flag-tagged S-phase kinase-associated protein 1 (Flag-Skp1), Cullin 1 (Cul1), and His-IKKβ were kindly gifted from Dr. Kazuhiro Iwai (Kyoto University). The baculoviruses were amplified using Sf9 cells (Thermo Fisher Scientific) to produce high-titer stocks.
Preparation of Purified Proteins
Petit-LUBAC composed of His-HOIP (ΔN)/HOIL-1L (ΔC)-Flag, and Full-LUBAC composed of His-HOIP/HOIL-1L-Flag, His-HOIP/Flag-SHARPIN or His-HOIP/HOIL-1L-Flag/Flag-SHARPIN, His-Ube1, His-TrCP1/Flag-Skp1 complex, Cul1/His-Rbx1 complex, and His-IKKβ, were prepared from expresSF+ cells (Protein Sciences Corporation, Meriden, CT) infected with the appropriate combinations of baculoviruses. GST-tagged ubiquitin (Ubi-GST), His-UbcH5c, and GST-IκBα were expressed in Escherichia coli Rosetta2 (DE3) (Merck KGaA).
The purification of recombinant proteins expressed in mammalian cells, baculovirus, and bacteria was performed using appropriate affinity resins, such as Ni-NTA Superflow (Qiagen, Hilden, Germany), Glutathione Sepharose 4B (GE Healthcare), and anti-Flag M2 affinity gel (Sigma-Aldrich, St. Louis, MO). Phosphorylated GST-IκBα (GST-p-IκBα) was prepared by phosphorylation of GST-IκBα with His-IKKβ, followed by purification using the Glutathione Sepharose 4B resin. Biotinylated-ubiquitin (Bio-Ubi) was prepared by ubiquitin (R&D Systems, Minneapolis, MN) and EZ-Link Sulfo-NHS-LC-LC-biotin (Thermo Fisher Scientific), 230 and 23 µM, respectively.
HTRF-Based LUBAC-Mediated Linear Ubiquitination Assay
Ubiquitination reactions were performed in 384-well plates (cat. 3656; Corning, Corning, NY). In HTS primary screening, 2 µL of each compound (0.75% DMSO) in reaction buffer (20 mM Tris-HCl [pH 7.5], 0.1% Triton X-100, 0.5 mM DTT [dithiothreitol], and 0.1% bovine serum albumin [Sigma-Aldrich]) was added to 4 µL of reaction buffer in 384-well plates using EDR-384UX (Biotec, Tokyo, Japan). Then, 4 µL of E1/E3 solution (37.5 nM Ube1 as E1, 150 nM Petit-LUBAC [complex of His-HOIP {ΔN} and HOIL-1L {ΔC}-Flag] as E3, 18.8 mM MgCl2, and 1.88 mM ATP in reaction buffer) was added and preincubated at room temperature (RT) for 60 min. The E1/E3 solution without E3 was used as a blank control. Five microliters of Ubi/E2 solution (1.2 µM Ubi-GST, 3.6 µM Bio-Ubi, and 7.2 µM Ubi, 375 nM UbcH5c as E2 in reaction buffer) was added and then incubated at RT for 12 min. After incubation, 30 µL of stop solution (45 mM ethylenediaminetetraacetic acid [EDTA] in Tris-buffered saline with 0.1% Tween-20 [TBST]) was added to stop the ubiquitination reactions.
HTRF assays were performed in white shallow 384-well plates (cat. 6008280; PerkinElmer, Waltham, MA). Two microliters of the ubiquitination reactions as described above was added to 6 µL of europium cryptate-conjugated streptavidin (SA-Eu; Cisbio Bioassays, Codolet, France) solution (3.75 nM SA-Eu, 30 mM EDTA, 533 mM KF, and 6.67% Blocking One in TBST) in 384-well plates. Then, 10 µL of d2-conjugated anti-GST mouse monoclonal antibody (GST-d2; Cisbio Bioassays) solution (12 nM GST-d2, 30 mM EDTA, 400 mM KF, and 5% Blocking One [Nacalai Tesque, Kyoto, Japan] in TBST) was added and incubated at RT for >60 min. After incubation, the HTRF signal (excitation at 337 nm, emission at both 655 and 616 nm) was read using a PARADIGM Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA). The HTRF ratio was calculated using the following equation: HTRF signal at 655 nm/HTRF signal at 616 nm × 10,000.
To monitor ubiquitin chain formation catalyzed by LUBAC, aliquots of the ubiquitination reaction products were mixed with LDS Sample Buffer (Thermo Fisher Scientific), separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) with NuPAGE gels (Thermo Fisher Scientific), and transferred to polyvinylidene difluoride (PVDF) membrane using a semidry transfer device (iBlot Gel Transfer Device; Thermo Fisher Scientific). Membranes were blocked in Blocking One and probed with indicated antibodies, and resulting bands were developed with ECL Plus (GE Healthcare) and imaged on an LAS-4000 imager (Fuji Film, Tokyo, Japan).
HTRF-Based p-IκBα Polyubiquitination Assay
The HTRF-based p-IκBα polyubiquitination assay differed from the primary screening in the following respects: (1) GST-p-IκBα was used instead of Ubi-GST, and (2) ubiquitination reactions were performed using 375 nM of the TrCP1/Skp1/Cul1/Rbx1 complex as E3 for 60 min.
NF-κB Luciferase Reporter Gene Assay in LUBAC-Overexpressing Cells
HEK293T/17 cells (human embryonic kidney cells expressing SV40 large T antigen; CRL-11268, ATCC, Manassas, VA) were seeded in a 225 cm2 flask in Dulbecco’s modified Eagle’s medium (high glucose) supplemented with 10% fetal bovine serum (Moregate Biotech, Bulimba, QLD, Australia) 1 day before transfection. Then, a total of 45 µg of the expression plasmid, including pGL4.32 (Promega, Madison, WI) as the NF-κB luciferase reporter plasmid, pcDNA3.1(+)/HOIL-1L-Flag, and pcDNA3.1(+)/His-HOIP, was cotransfected using 45 µL of Lipofectamine LTX Reagent (Thermo Fisher Scientific) according to the manufacturer’s manual. After 5 h, transfected cells were trypsinized and cryopreserved in Cellbanker 1 (Nippon Zenyaku Kogyo, Fukushima, Japan) at −80 °C and used in subsequent studies. Cryopreserved cells were seeded in 96-well plates, and compounds were added to the plates at the same time. After 7–19 h of incubation, 100 µL of ONE-Glo Reagent (Promega) for luciferase activity measurement, or CellTiter-Glo Reagent (Promega) for quantifying cytotoxicity, was added. The luciferase signal was measured using a LEADseeker imaging system (GE Healthcare).
NF-κB Luciferase Reporter Gene Assay in NEMO-(Ubi)3-Overexpressing Cells
A total of 45 µg of expression plasmids, including pGL4.32, pcDNA3.1(+)/HA-NEMO, pcDNA3.1(+)/HA-NEMO-(Ubi)3, and pcDNA3.1(+), was used for transfection. The following steps were performed as described in the section on NF-κB luciferase reporter gene assay in LUBAC-overexpressing cells.
LC/MS Aalysis of Hit Compounds in Buffer
Liquid chromatography–mass spectrometry (LC/MS) analysis was performed on an 1100 Series HPLC (Agilent Technologies, Santa Clara, CA) and Acquity SQ Detector (Waters Corporation, Milford, MA) with a C18 reversed-phase column (BetaBasic-18, 50 × 2.1 mm; Thermo Fisher Scientific). Mobile phase A consisted of water with 0.1% trifluoroacetic acid, and mobile phase B consisted of acetonitrile with 0.1% trifluoroacetic acid. The LC gradient was from 5% B to 95% B over 9 min and was held at 100% B for 2 min. The flow rate was 0.3 mL/min. Data analysis was operated using the chromatogram on 214 nm, and assignment of chemical species was conducted using MS scan (m/z 200–800) in positive-ion detection mode.
A 4 mM DMSO solution of JTP-1048196 (6 µL) and “Tris buffer” (20 mM Tris-HCl [pH 7.5]; 154 µL) was mixed to make a 0.15 mM JTP-1048196 solution in 3.75% DMSO–Tris buffer, with or without 0.5 mM DTT. The first analysis was performed immediately after mixing the two solutions (0 h); the second analysis was done after the indicated time, as shown in Supplemental Table S1 .
Cell Culture
HEK293T and HeLa cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 100 IU/mL penicillin G, and 100 μg/mL streptomycin at 37 °C under a 5% CO2 atmosphere.
SDS-PAGE and Western Blotting
Samples were separated by SDS-PAGE and transferred to PVDF membranes. After blocking, the membrane was incubated with the appropriate primary antibodies, followed by an incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies. The chemiluminescent images were obtained with an LAS4000 imaging analyzer (GE Healthcare) or a Fusion Solo S imaging system (Vilber, Marne-la-Vallée, France).
Antibodies
Antibodies were purchased or supplied as indicated below: anti-Ubi mouse monoclonal IgG (P4D1; Santa Cruz Biotechnology, Dallas, TX), HRP-conjugated sheep polyclonal antibody anti-mouse IgG (GE Healthcare), and HRP-conjugated anti-GST goat polyclonal antibody (GE Healthcare). Moreover, p-p105 (no. 4806), p105 (no. 13586;), p-p65 (no. 3033), p65 (no. 8242), and p-IKKα/β (no. 2697) were obtained from Cell Signaling Technology (Danvers, MA). IKKα/β (H-470, sc-7607) was purchased from Santa Cruz Biotechnology. Linear ubiquitin (LUB9, MABS451; Merck KGaA), tubulin (CLT9002; Cedarlane, Ontario, Canada), and HA (Roche, Basel, Switzerland) were also used.
Quantitative PCR
Cell lysis, reverse transcription, and quantitative PCR (qPCR) were performed with a SuperPrep Cell Lysis RT Kit for qPCR (TOYOBO, Osaka, Japan) and Power SYBR Green PCR Master Mix (Thermo Fisher Scientific), according to the manufacturers’ instructions. Quantitative real-time PCR was performed with a Step-One-Plus PCR system (Thermo Fisher Scientific) by the ΔΔCT method, using the following oligonucleotides: human ICAM1 sense, 5′-GTGGTAGCAGCCGCAGT-3′, and human ICAM1 anti-sense, 5′-TTCGGTTTCATGGGGGT-3′; human TNF-α sense, 5′-GCCGCATCGCCGTCTCCTAC-3′, and human TNF-α anti-sense, 5′-CCTCAGCCCCCTCTGGGGTC-3′; and human GAPDH sense, 5′-AGCAACAGGGTGGTG GAC-3′, and human GAPDH anti-sense, 5′-GTGTGGT GGGGGACTGAG-3′.
Data Analyses
The inhibitory activity (percent) of compounds was calculated using the following equation: (1 – [average of compounds well − average of blank well]/[average of control well − average of blank well]) × 100.
Data analyses were performed using Prism, version 6.01, from GraphPad Software Inc. (San Diego, CA). Concentration–response curves were fitted using nonlinear regression analysis.
The Z′ factor was determined using the following equation: 18 Z′ = 1 − (3σc+ + 3σC−)/│µC+ − µC−│, where µC+ and µC− are the means of the control well signal and blank well signal, respectively, and σc+ and σC− are the standard deviations of the corresponding signals. In qPCR analyses, one-way ANOVA followed by a post-hoc Tukey HSD test was performed using KaleidaGraph software. For all tests, a p value of less than 0.05 was considered statistically significant.
Results
Development of the HTRF-Based Cell-Free LUBAC-Mediated Linear Ubiquitination Assay
Recombinant Ube1 as E1, UbcH5c as E2, and Full-LUBAC (complex of His-HOIP and HOIL-1L-Flag) or Petit-LUBAC (complex of His-HOIP [ΔN] and HOIL-1L [ΔC]-Flag) as E3 for the ubiquitination assay were produced by bacterial, baculovirus, or mammalian expression systems. Each activity of the purified protein was confirmed as described below. Ubiquitin chain formation catalyzed by Full-LUBAC was monitored by SDS-PAGE followed by Western blot analysis of a reaction mixture containing E1, E2, LUBAC, ubiquitin, and ATP, referred to as the “complete reaction mix.” Reactions lacking any of these components were compared with the complete reaction mix to confirm the selectivity against LUBAC. As a result, ubiquitin chain elongation was detected only in the complete reaction mix ( Suppl. Fig. S2 ).
To screen LUBAC inhibitors from our compound library, we developed a high-throughput assay to detect LUBAC-mediated linear ubiquitinations using HTRF technology with Petit-LUBAC (complex of His-HOIP [ΔN] and HOIL-1L [ΔC]-Flag). We utilized the following labeled ubiquitins: Ubi-GST (C-terminus GST-tagged ubiquitin) as the first incorporated ubiquitin, and Bio-Ubi and nonlabeled ubiquitin, which were randomly incorporated into the polyubiquitin chain. In principle, during ubiquitin chain elongation by LUBAC, if the first incorporated ubiquitin was Ubi-GST and subsequent ubiquitins were Bio-Ubi, the polyubiquitin chain was detected by the energy transfer between the donor SA-Eu and the acceptor GST-d2 (d2-conjugated anti-GST antibody). The excitation of the donor at 360 nm, followed by its emission at 620 nm, results in an energy transfer to the proximal acceptor via FRET. This energy transfer occurs when SA-Eu and GST-d2 are in proximity to the polyubiquitin chains. This allows the ratiometric measurement of acceptor emission at 665 nm over the 620 nm donor emission, constituting the HTRF ratio ( Fig. 1A ).
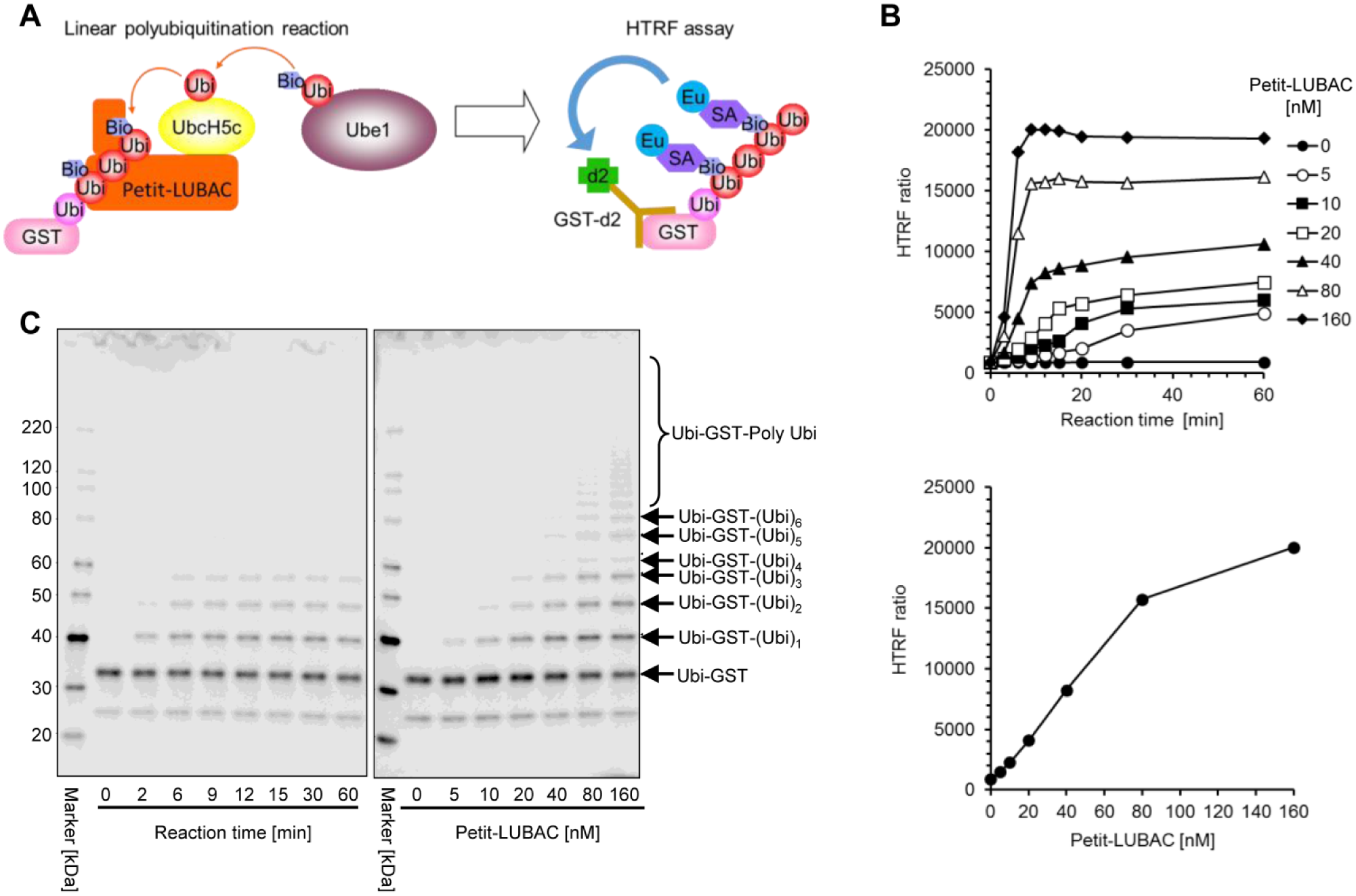
Development of the HTRF-based LUBAC-mediated linear polyubiquitination assay. (
To optimize the assay conditions, we carried out titrations of enzyme (E1 and E2), Ubi-GST, the ratio of Bio-Ubi to nonlabeled ubiquitin, and the dilution factor (amount of ubiquitination reactions added to the HTRF reaction) ( Suppl. Fig. S3 ). A time-course experiment was also conducted to determine the optimal evaluation time and concentration of Petit-LUBAC for compound screening. The HTRF ratio was found to be proportional to the concentration of Petit-LUBAC up to 80 nM at 12 min of reaction time ( Fig. 1B ). The HTRF ratio also correlated with the length of formed polyubiquitin chains, as demonstrated by Western blot analysis of reaction products using the antibody specific to GST ( Fig. 1C ). On the basis of these results, test compounds were evaluated with 40 nM Petit-LUBAC at a 12 min reaction time. We confirmed that at least 5% DMSO does not affect the cell-free assay.
As a counterassay to the HTRF-based LUBAC-mediated linear ubiquitination assay, we developed an HTRF-based p-IκBα polyubiquitination assay to exclude nonspecific compounds. The assay components were the same as those of the LUBAC-mediated ubiquitination assay with the exception that E3 was the TrCP1/Skp1/Cul1/Rbx1 complex and the substrate was p-IκBα ( Suppl. Fig. S4A ). Titrations of enzyme (E1 and E2), the ratio of Bio-Ubi to nonlabeled ubiquitin, and the dilution factor were carried out as described in the LUBAC-mediated linear ubiquitination assay. A time-course experiment was also conducted to determine the optimal evaluation time and concentration of E3 for compound screening. The HTRF ratio was found to be proportional to the concentration of E3 up to 200 nM at 60 min of reaction time ( Suppl. Fig. S4B ). It also correlated with the length of formed polyubiquitin chains, as demonstrated by Western blot analysis of the reaction products using the antibody specific to GST ( Suppl. Fig. S4C ). On the basis of these results, test compounds were evaluated with 100 nM E3 at 60 min of reaction time. We confirmed that at least 5% DMSO does not affect the cell-free assay.
Development of the Cell-Based LUBAC Assay Using the NF-κB Luciferase Reporter Gene Assay
We developed a cell-based NF-κB luciferase reporter gene assay in LUBAC-overexpressing cells to evaluate the cellular activity of compounds, based on the report that transient expression of LUBAC in mammalian cells activates the NF-κB signal via linear ubiquitination of endogenous NEMO. 8 Thus, compounds inhibiting intracellular LUBAC may suppress luciferase activity induced by NF-κB activation in LUBAC-overexpressing cells.
In this study, HEK293T cells were transiently transfected with the NF-κB luciferase reporter plasmid, and with pcDNA3.1(+)/HOIL-1L-Flag and pcDNA3.1(+)/His-HOIP as LUBAC expression plasmids. A significant increase in luciferase activity was detected only in HOIL-1L and HOIP coexpressing cells at 24 h after transfection ( Fig. 2A ). To determine the optimal evaluation time for compound screening, we measured luciferase activity in the cells. Luciferase activity gradually increased up to 19 h after reseeding the cryopreserved cells, which were stocked at 5 h after transfection ( Fig. 2B ). On the basis of these results, test compounds were added into cell cultures immediately after reseeding the cryopreserved cells, and the activity of each compound on luciferase activity and cytotoxicity was evaluated after 19 h.
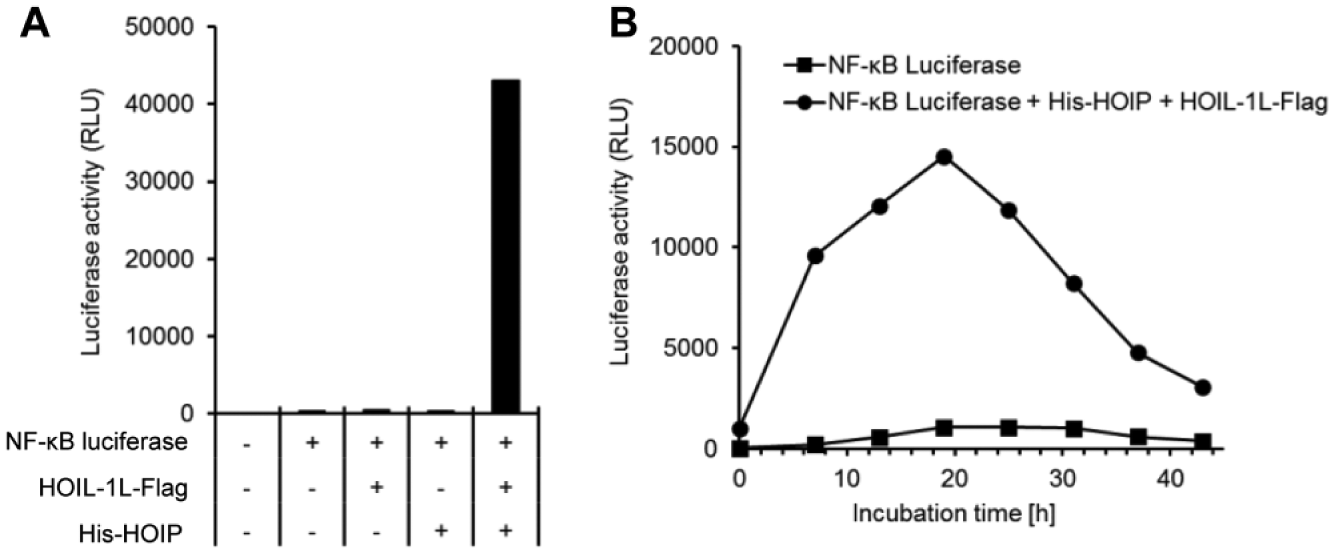
Development of the NF-κB luciferase reporter gene assay in LUBAC-overexpressing cells. (
In addition, we developed a counterassay to the cell-based LUBAC assay using the NF-κB luciferase reporter gene assay. The counterassay was set up in order to exclude compounds inhibiting downstream effectors from the linear ubiquitination of NEMO by LUBAC. To this end, we generated a fusion gene construct coding HA-NEMO fused with linear ubiquitin composed of three uncleavable ubiquitins at its C-terminus (HA-NEMO-Ubi[G76V]-Ubi[G76V]-Ubi[G75,76A]) end, referred to as HA-NEMO-(Ubi)3. This was based on the report that transient expression of NEMO, fused with at least two linear ubiquitins at the C-terminus of the protein in HEK293T cells, activates sufficient NF-κB activity. 19
In the present study, HEK293T cells were transiently transfected with the NF-κB luciferase reporter plasmid, and with pcDNA3.1(+)/HA-NEMO or pcDNA3.1(+)/HA-NEMO-(Ubi)3 as expression plasmids. After a 5 h transfection, cells were cryopreserved and cultured after thawing for up to 43 h. Luciferase activity in the cells was more than three times higher and reached its peak at about 7 h after reseeding, which was a shorter culture time than that observed in LUBAC-overexpressing cells ( Suppl. Fig. S5 ). On the basis of these results, test compounds were added immediately after reseeding the cryopreserved cells, and the activity of the compounds on luciferase activity and cytotoxicity was evaluated after 7 h.
HTS for LUBAC-Selective Inhibitors
First, approximately 250,000 chemical compounds from the library of Japan Tobacco Inc. were initially screened at a single concentration of 30 µM using the HTRF-based LUBAC-mediated ubiquitination assay with Petit-LUBAC (complex of His-HOIP [ΔN] and HOIL-1L [ΔC]-Flag). The primary screen was accomplished with high quality, as observed by robust Z′ factors and HTRF ratios. The Z′ factor is an indicator of screening system reliability and is judged to be sufficient when it is larger than 0.5. 18 In this study, the Z′ factor was 0.91 ± 0.020 (mean ± SD; Fig. 3A ). In addition, the HTRF ratio in the blank, control, and positive control (30 mM NaCl) wells of each plate was stable ( Fig. 3B ). NaCl was shown to inhibit LUBAC activity in the HTRF-based LUBAC-mediated ubiquitination assay and was used as a positive control. The hit criterion was defined as more than 15% inhibition, and 416 compounds were selected as the first hit compounds with a hit rate of 0.17% ( Fig. 3C ). The first hit compounds were retested with the same assay, and 170 compounds showing more than 30% inhibition were identified as confirmed hit compounds.
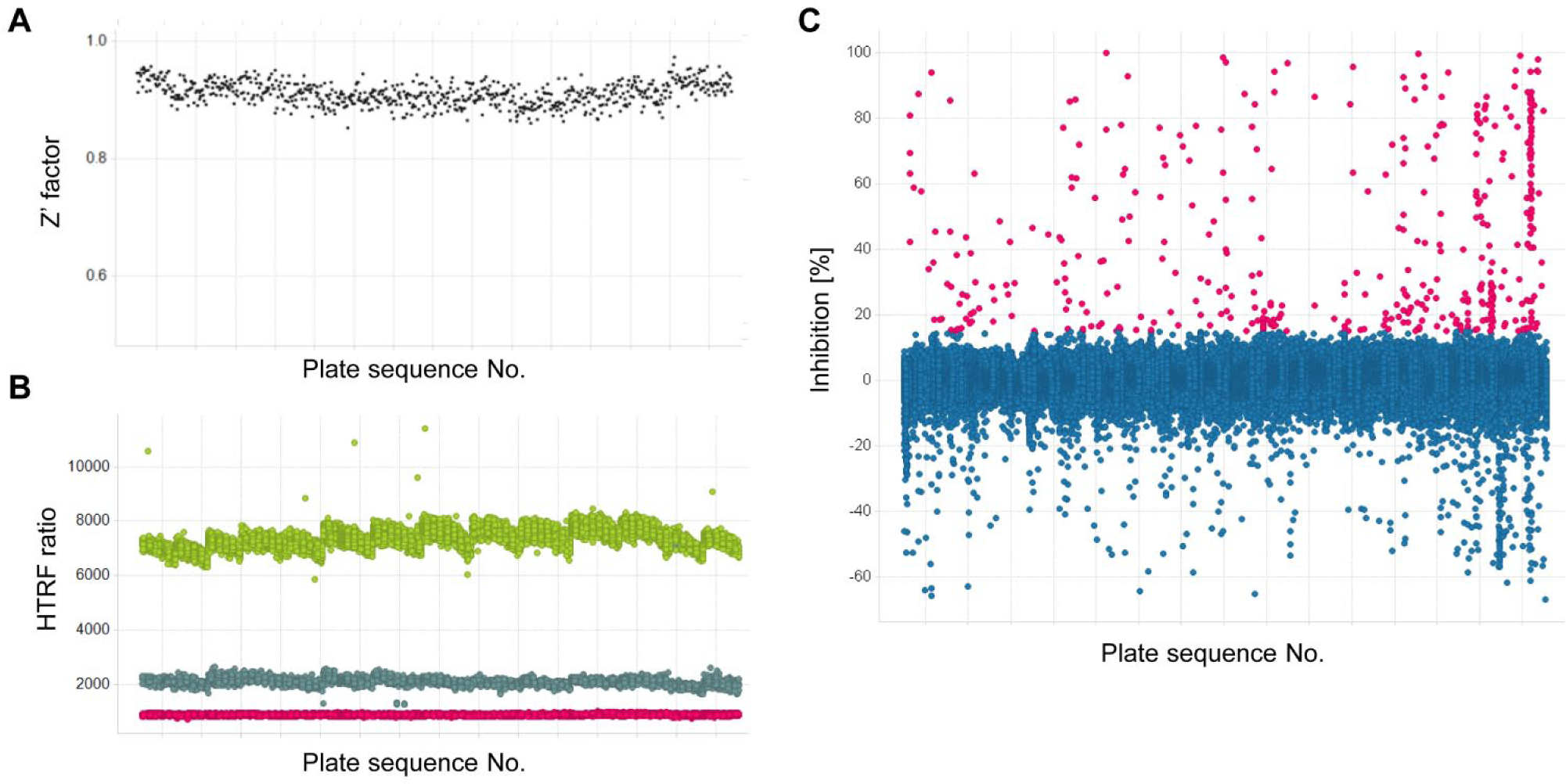
Results of primary screening. Approximately 25,000 chemical compounds in the library on 723 plates (384-well plate) were screened at final concentrations of 30 µM using the HTRF-based LUBAC-mediated linear ubiquitination assay with Petit-LUBAC (complex of His-HOIP [ΔN] and HOIL-1L [ΔC]-Flag). (
We next conducted the HTRF-based LUBAC-mediated ubiquitination assay using Petit-LUBAC (complex of His-HOIP [ΔN] and HOIL-1L [ΔC]-Flag), as well as its counterassay, to evaluate the selectivity of the 170 confirmed hit compounds. Concentration–response curves were generated, and 22 compounds showed a concentration-dependent inhibition with IC50 values of less than 40 μM in the LUBAC-mediated ubiquitination assay, and more than fourfold selectivity against the counterassay using the E3 (TrCP1/Skp1/Cul1/Rbx1 complex)-mediated p-IκBα polyubiquitination assay. A concentration–response curve of a representative compound (final hit compound JTP-1048196) is shown in Figure 4A .
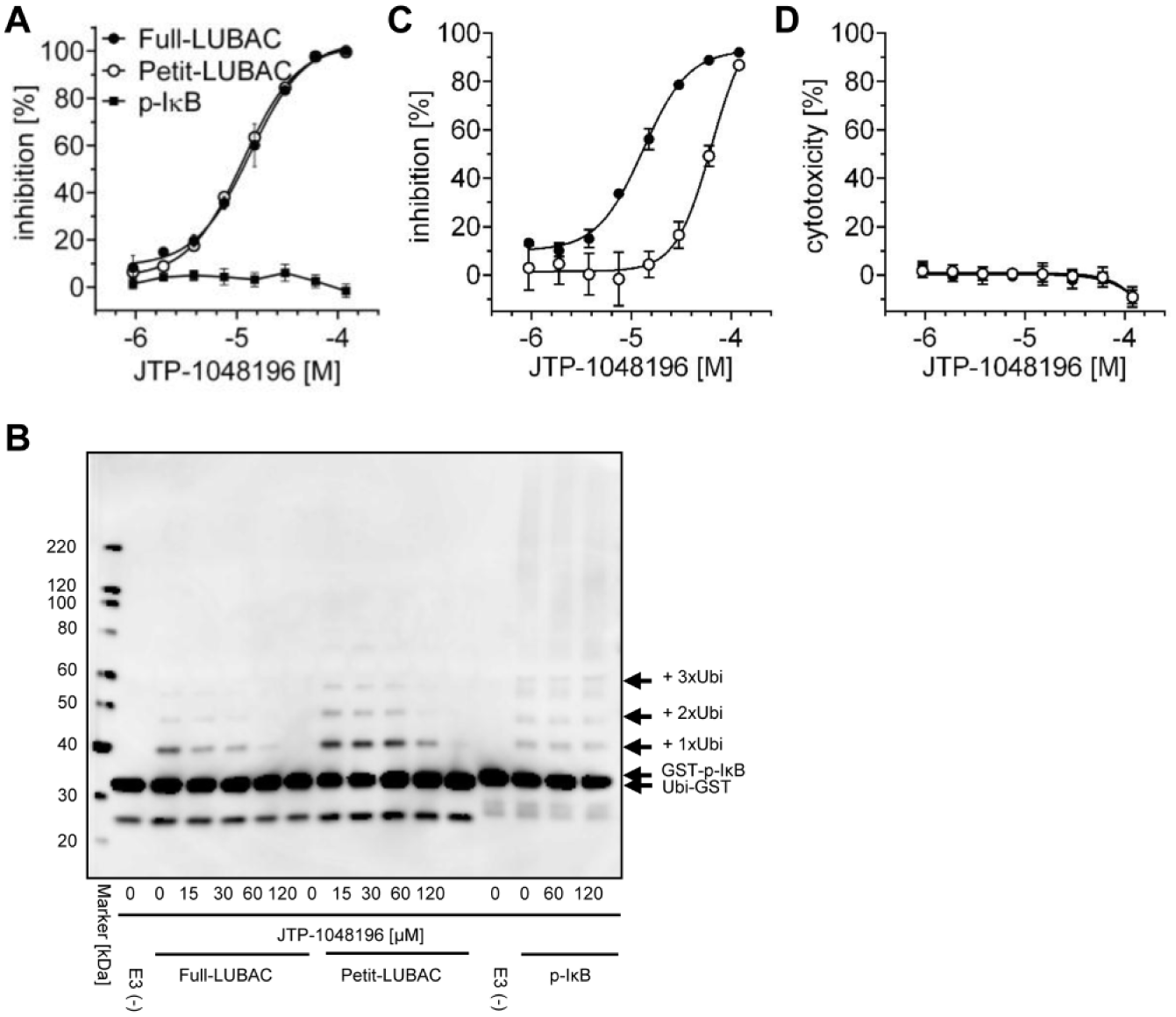
The activity profile of JTP-1048196. (
We then confirmed that 22 compounds inhibited the Full- and Petit-LUBAC-mediated linear ubiquitination reaction but did not inhibit the E3 (TrCP1/Skp1/Cul1/Rbx1 complex)-mediated p-IκBα polyubiquitination reaction. This was determined by polyubiquitin chain formation, which was visualized with Western blotting using the anti-GST antibody. The results of Western blot analysis correlated well with those of HTRF-based assay. Representative data using the final hit compound JTP-1048196 are shown in Figure 4B . We next conducted the cell-based LUBAC assay, and concentration–response curves were generated for the 22 compounds. We selected 13 compounds that showed a concentration-dependent inhibition with IC50 values less than 40 μM and less cytotoxicity. These 13 compounds were classified into four categories on the basis of the chemical structure.
Furthermore, we performed the counterassay to the cell-based LUBAC assay to eliminate compounds inhibiting downstream effectors from linearly ubiquitinated NEMO using NEMO-(Ubi)3-overexpressing cells. All four compounds in the same category were selected as final hit compounds because they met the hit criterion, which included IC50 values for LUBAC less than 40 μM, with more than fourfold selectivity against the counterassay. JTP-1048196 was selected as a representative final hit compound from the category ( Fig. 4C ). JTP-1048196 was evaluated at time points similar to those of the NEMO-(Ubi)3 assay (7 h incubation), and the IC50 values were found to be 13.3 ± 3.1 µM for LUBAC and 73.8 ± 2.6 µM for NEMO at 7 h incubation (means ± SEM, n = 3). JTP-1048196 did not show cytotoxicity ( Fig. 4D ), and the activity profile of the compound is summarized in Table 1 .
Activity Profiles of JTP-1048196 and JTP-0819958 for the HTRF-Based and Cell-Based Assay.
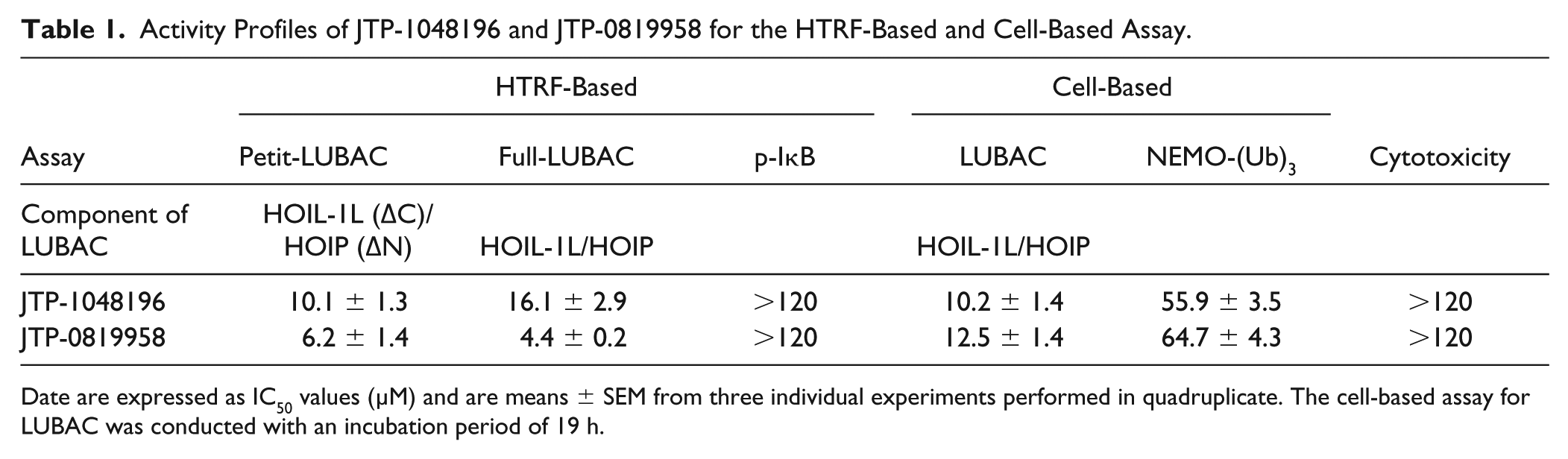
Date are expressed as IC50 values (µM) and are means ± SEM from three individual experiments performed in quadruplicate. The cell-based assay for LUBAC was conducted with an incubation period of 19 h.
Characterization of Final Hit Compounds with a Lactone Moiety
We investigated the behavior of the hit compounds in the reaction buffer of the HTRF-based LUBAC-mediated ubiquitination assay because all four final hit compounds in the category contained a chemically reactive lactone structure. They were therefore assumed to be isomerized in the reaction buffer. LC/MS analysis revealed that a lactone structure of JTP-1048196 was transformed to give JTP-0819958 time dependently in the reaction buffer ( Fig. 5A , Suppl. Table S1 ). Consistent with LC/MS analysis, the inhibitory activity of JTP-1048196 to Petit-LUBAC increased gradually with a longer preincubation time in the reaction buffer, and the IC50 value decreased from 26 μM for 1 h to 3.9 μM for 24 h preincubation ( Fig. 5B ).
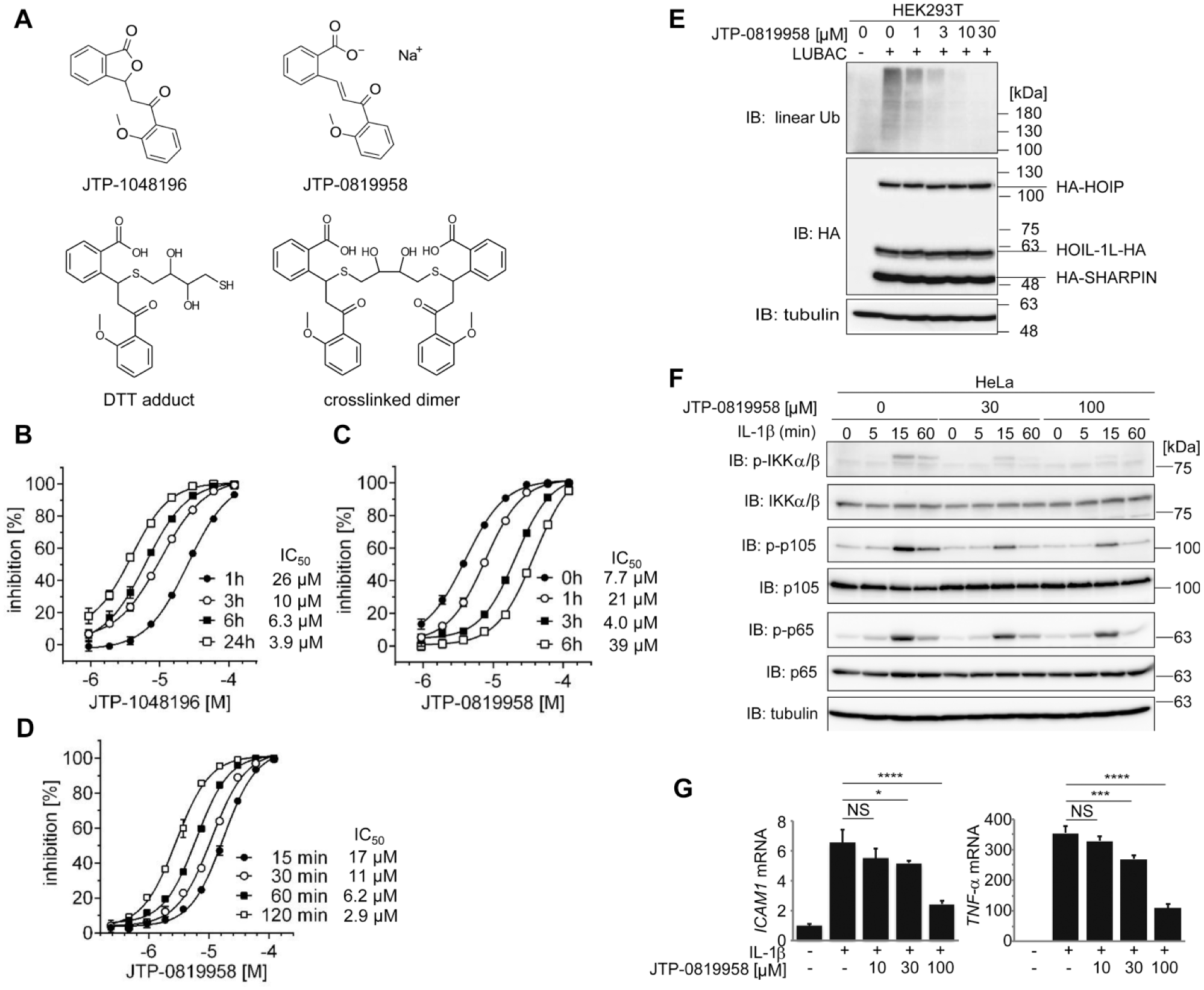
A lactone structure of JTP-1048196 is transformed to reactive JTP-0819958. (
Because JTP-0819958 possesses a thiol-reactive α,β-unsaturated carbonyl moiety, we examined whether the reactive moiety reacts with DTT and affects the activity of Petit-LUBAC. The inhibitory activity of JTP-0819958 decreased gradually with a longer preincubation time in the reaction buffer containing DTT, and the IC50 value increased from 4.0 μM for 0 h to 39 μM for 6 h preincubation with DTT ( Fig. 5C ). LC/MS analysis confirmed that JTP-0819958 reacted with DTT to give DTT adducts or crosslinked dimers in the reaction buffer containing DTT ( Suppl. Table S1 ). Next, we examined the time-dependent of inhibition of JTP-0819958 with the LUBAC-mediated linear ubiquitination assay. The inhibitory activity was found to be increased with a longer incubation time with Petit-LUBAC ( Fig. 5D ). The results indicate that JTP-0819958 is a covalent inhibitor of LUBAC. The activity profile of JTP-0819958 is summarized in Table 1 .
Finally, to examine the inhibitory effects of JTP-0819958 on LUBAC activity, we analyzed intracellular linear ubiquitination. The coexpression of the LUBAC subunits in HEK293T cells increased the intracellular amounts of linear polyubiquitin, whereas JTP-0819958 dose dependently suppressed the production of intracellular linear polyubiquitin ( Fig. 5E ). Moreover, IL-1β-stimulated NF-κB activation, demonstrated by the phosphorylation of IKKα/β, p105, and p65, was suppressed by JTP-0819958 in HeLa cells ( Fig. 5F ), and the expression of NF-κB target genes such as ICAM1 and TNF-α was reduced by JTP-0819958 in IL-1β-treated HeLa cells ( Fig. 5G ). Our results suggested that the lactone structure of JTP-1048196 was transformed to a reactive α,β-unsaturated carbonyl moiety, which reacts with the cysteine residue of LUBAC, leading to its covalent inhibition in vitro and cellular levels.
Discussion
In this study, we developed a novel assay that directly detected the linear polyubiquitination by LUBAC, and adapted this assay into a robust HTS format. In addition to this cell-free enzyme assay, the cell-based NF-κB luciferase reporter gene assay was used to further demonstrate the enzyme activity of LUBAC in a cellular environment. In the HTS campaign, it is necessary to exclude nonspecific compounds. Compounds inhibiting E1 or E2 in the HTRF-based LUBAC-mediated ubiquitination assay were eliminated by the counterassay using the same E1, E2, and different E3. In addition, compounds inhibiting any downstream components of the NF-κB signal cascade from linearly ubiquitinated NEMO by LUBAC were eliminated by the counterassay using NEMO-(Ubi)3-overexpressing cells in which NF-κB signal cascades from linearly ubiquitinated NEMO were activated. Thus, the combination of the cell-free or cell-based assay and each counterassay served to exclude nonspecific compounds. Using the combined platform of the HTRF-based assay and the cell-based NF-κB luciferase reporter gene assay, we successfully identified a chemical series that inhibits LUBAC activity both biochemically and in the cellular context. The platform we described in this study may provide a powerful tool for identifying inhibitors amenable to further optimization for therapeutic purposes.
Various methods exist to label biological molecules for the HTRF assay. Direct labeling is achieved through chemical reactions in order to attach fluorescent probes to target molecules. Indirect labeling generally involves the use of secondary molecules that are labeled with fluorescent probes. The most frequently used secondary molecules are streptavidin or anti-epitope tag antibodies, such as anti-GST, anti-FLAG, anti-His, or anti-HA. The indirect labeling approach for the HTRF assay is attractive because a variety of optimized labeled secondary reagents are available. In addition, the covalent modification of ubiquitin is avoided by direct labeling, which may affect biological function. 20
The previously reported TR-FRET-based LUBAC-mediated ubiquitination assay utilized the direct labeling method (terbium cryptate-labeled ubiquitin and fluorescein-labeled ubiquitin). 17 In developing the HTRF-based LUBAC-mediated ubiquitination assay that we used here, we chose an indirect method for labeling ubiquitin with europium or d2, taking advantage of available labeled secondary reagents (europium cryptate-labeled streptavidin and d2-labeled anti-GST antibody). Moreover, the amplification of the HTRF ratio was achieved by the strong affinity of streptavidin to biotin. These improvements for the HTRF assay resulted in high ubiquitination efficiency and sensitivity for detecting the linear polyubiquitin chain.
Our HTRF-based LUBAC-mediated ubiquitination assay has more favorable characteristics than the previously reported TR-FRET-based assay has. 17 The previously reported assay mainly detected the longer length of linear ubiquitin chains, whereas our assay accurately measured the shorter length of linear ubiquitin chains. In our experiment, an approximately linear polyubiquitin composed of six ubiquitins was detected by Western blot analysis under optimal assay conditions (12 min of reaction time with 40 nM Petit-LUBAC). Under physiological conditions, more importantly, it is suggested that linear ubiquitin chains are tightly regulated in their production and/or are forming shorter chains preferentially owing to their rapid cleavage in the cellular cytosol. 19 It has also been demonstrated that linear di-ubiquitins are sufficient to activate the IKK complex in vitro, and to trigger maximal NF-κB activation in cells. 19 Consequently, our HTRF-based LUBAC-mediated ubiquitination assay should be suitable for detecting physiological LUBAC activity.
Preliminary experiments showed that JTP-0819958 inhibited the linear polyubiquitination activity by the HOIL-1L/HOIP complex, the HOIL-1L/HOIP/SHARPIN complex, and the HOIP/SHARPIN complex equally, with IC50 values of 4.4 ± 0.2, 3.5 ± 0.3, and 3.7 ± 0.4 μM (means ± SEM, n = 3), respectively. HOIP is the catalytic core of LUBAC and the common component among these three complexes used in this study. Furthermore, the inhibitory effects of JTP-0819958 on LUBAC-mediated linear ubiquitin generation and inflammatory cytokine-induced NF-κB activation were confirmed in cellular levels. Taken together, we speculated that HOIP may be the target of the compound. In addition, although all hit compounds that we identified have reactive α,β-unsaturated carbonyl groups, they have no inhibitory effect against the counterassay using the same E1, E2, and different E3 (TrCP1/Skp1/Cul1/Rbx1 complex). Thus, these compounds were presumed to selectively react with cysteine residues of the HOIP catalytic core.
Although further investigation is needed to elucidate the precise mechanism of action of these compounds as potential covalent inhibitors, covalent inhibitor–target interactions offer significant pharmacological advantages, including high biochemical efficiency and prolonged duration of action, resulting in lower doses and reduced off-target effects.21,22 JTP-0819958 may become a useful research tool for elucidating the molecular mechanisms of in vitro LUBAC-mediated polyubiquitination and should promote further research for better understanding the molecular mechanisms of LUBAC-mediated ubiquitination.
Supplemental Material
Revised_supplemental_date – Supplemental material for High-Throughput Screening for Linear Ubiquitin Chain Assembly Complex (LUBAC) Selective Inhibitors Using Homogenous Time-Resolved Fluorescence (HTRF)-Based Assay System
Supplemental material, Revised_supplemental_date for High-Throughput Screening for Linear Ubiquitin Chain Assembly Complex (LUBAC) Selective Inhibitors Using Homogenous Time-Resolved Fluorescence (HTRF)-Based Assay System by Ken Katsuya, Yuji Hori, Daisuke Oikawa, Tomohisa Yamamoto, Kayo Umetani, Toshiki Urashima, Tomomi Kinoshita, Kumiko Ayukawa, Fuminori Tokunaga and Masahiro Tamaru in SLAS Discovery
Footnotes
Acknowledgements
We thank Dr. Kazuhiro Iwai for providing recombinant baculoviruses encoding His-TrCP1, Flag-Skp1, Cul1, and His-IKKβ, and for useful discussions. We also thank Shin-ya Sasaki (Japan Tobacco Inc.) for LC/MS analysis, Kiyoto Nagahara (Japan Tobacco Inc.) for compound synthesis, and Kazuki Hanada (Japan Tobacco Inc.) for helpful discussions.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.