
Abstract
Hantaviruses cause hemorrhagic fever with renal syndrome (HFRS) and hantavirus cardiopulmonary syndrome (HCPS), which infects more than 200,000 people worldwide. Sin Nombre virus (SNV) and Andes virus (ANDV) cause the most severe form of HCPS, with case fatality ratios of 30%–40%. There are no specific therapies or vaccines for SNV. Using high-throughput flow cytometry, we screened the Prestwick Chemical Library for small-molecule inhibitors of the binding interaction between UV-inactivated and fluorescently labeled SNVR18 particles, and decay-accelerating factor (DAF) expressed on Tanoue B cells. Eight confirmed hit compounds from the primary screen were investigated further in secondary screens that included infection inhibition, cytotoxicity, and probe interference. Antimycin emerged as a bona fide hit compound that inhibited cellular infection of the major HCPS (SNV)- and HCPS (Hantaan)-causing viruses. Confirming our assay’s ability to detect active compounds, orthogonal testing of the hit compound showed that antimycin binds directly to the virus particle and blocks recapitulation of physiologic integrin activation caused by SNV binding to the integrin PSI domain.
Keywords
Introduction
Hantaviruses are an important group of emerging pathogenic viruses and represent a paradigm for zoonotic airborne transmission. Sin Nombre virus (SNV) is a Category A pathogen that causes hantavirus cardiopulmonary syndrome (HCPS) with case fatality rates of 30%–50%. 1 Tissue tropism involves the vascular endothelium of the heart, kidney, lung, and lymphoid organs, where dysregulation of the endothelial barrier function, edema, and focal hyaline membranes are central features.2–4 There is no approved, effective therapy, and treatment of the disease is supportive, including the use of extracorporeal membrane oxygenation (ECMO). 5 The ability of a virus to break the species barrier critically depends on its capacity to enter human cells efficiently. Inhibiting viral entry allows blocking the pathogen before it can take control of the host cell and represents a promising strategy for therapeutic antiviral intervention.
The hantavirus genome comprises three segments of negative-sense ssRNA, which are coated with multiple copies of the nucleoprotein. A lipid bilayer membrane envelope in which two glycoproteins, Gn and Gc, are localized covers the virus. 4 These envelope glycoproteins are necessary for virus attachment to entry receptors and fusing with endocytic vesicles once inside the host cell. Pathogenic hantaviruses, including Andes (ANDV), SNV, Hantaan (HTNV), Puumala (PUUV), Seoul Virus, and New York-1 (NY-1), have been reported to use αvβ3 and αIlbβ3 integrins for host cell entry that leads to productive infection.6,7 The hantavirus engages the PSI domain of the β3 integrin subunit of low-affinity conformers of αvβ3 and αIIbβ3 integrins, to enter host cells. 6 The effect of hantavirus engagement of the PSI domain on integrin activity is not well understood. One long-held view is that hantaviruses bind to inactive integrins and maintain them in an inactive state. 7 However, recent single-molecule atomic force microscopy experiments from our lab have shown that SNV engagement of the integrin PSI domain induces higher affinity for its cognate RGD ligands. We further demonstrated that bent conformation low-affinity β3 integrins interact in cis with the G protein–coupled receptor (GPCR) P2Y2R via an RGD sequence in the first extracellular loop of P2Y2R. 8 As a result, hantavirus engagement of the integrin–P2Y2R complex stimulates a force-dependent integrin outside-in signaling cascade. Outside-in signaling requires a surface-anchored ligand, which can support traction force upon its exertion. 9 The cis binding to P2Y2R provides the requisite fixed scaffold for outside-in signaling. In this way, the physiologically activated (inside-out) and extended conformation integrin αIlbβ3 is recognized by PAC-1. 8 PAC-1 is an immunoglobulin M (IgM) antibody that selectively binds to the fibrinogen binding site of activated αIIbβ3. 10 Others have shown that pathogenic hantaviruses also recognize the glycosylphosphatidyl-inositol-linked protein, CD55/decay-accelerating factor (DAF), which they use as an effector for integrin activation during cell entry.11,12 DAF is a 70 kDa member of the complement system, which is often exploited by various pathogens to gain entry into host cells.13,14 Although the mechanism that links the interaction of hantaviruses with DAF and integrin-mediated cell entry is currently not well understood, it is known that cross-linking of DAF by proteins or viruses stimulates cell signaling cascades upstream of integrin activating pathways.13,15
In this study, we tested whether the combination of targeting DAF binding inhibitors in primary screens and the PSI domain in subsequent tests could be used to identify hits and their molecular mechanism of action. As an effector for cellular infection by multiple pathogens, DAF is an attractive target for discovery campaigns of small molecules that inhibit binding between DAF and its pathogenic ligands. DAF is expressed in a wide range of cells, including Tanoue B suspension cells, which are amenable to homogeneous flow cytometry assays.12,16 Homogeneous flow cytometry uses small sample volumes and allows the resolution of free and bound virus, and obviates a wash step. A high signal-to-background ratio of individual cells in suspension is possible compared with adherent cells in the virus or antibody-based assays. Finally, flow cytometry is readily amenable to multiplexing.
Accordingly, we developed a flow cytometry–based high-throughput screen (HTS) assay for small-molecule inhibitors of UV-inactivated and fluorescently labeled SNV (SNVR18), and DAF expressed in β3 integrin-null Tanoue B cells. 16 We conducted a primary HTS campaign of the Prestwick Chemical Library (PCL). We tested candidate compounds in orthogonal screens for probe interference, as well as inhibition of infection and cytotoxicity. To validate a potential mechanism of action of our hit compound, antimycin, we assayed the compound for its ability to interfere with SNV-induced integrin signaling cascade. We first applied the cAMP Difference Detector in situ (cADDis) BacMam fluorescent biosensor kit 17 to test the effect of antimycin on SNV-induced cyclic AMP production in CHO-A24 BacMam–transformed cells. Second, we tested the effect of antimycin on PAC-1 staining of SNV-activated cells. 8
Materials and Methods
Materials
1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine-N-(carboxyfluorescein) (fluorescein PE) and dioleoyl-
Cell Culture
Tanoue B cells and Vero E6 cells were maintained in either sterile filtered RPMI media or minimum essential media (MEM). All media contained 10% heat-inactivated fetal bovine serum (FBS), 100 units/mL penicillin, 100 µg/mL streptomycin, 10 mM HEPES, pH 7.4, 20 µg/mL ciprofloxacin, and 2 mM
Production of SNV
Stocks of SNV particles were propagated in Vero E6 cells as previously described. 19 The typical yield is ≈1010 particles/mL in 15 mL aliquots. The ratio of particle to infectious unit is 104 to 1. 16 The binding assays on which the HTS screen is based do not distinguish between infectious and noninfectious particles. The 15 mL stocks are concentrated to 1.5 mL. The virions are then rendered noninfectious by a 15 s dose of UV light at 254 nm as described. 19 The neutralization of irradiated supernatants is confirmed by a focus reduction assay before transfer, with documentation from the BSL-3 laboratory as required by UNM Biosafety. The structural integrity of UV-irradiated SNV is checked by immunoblotting of key protein components (i.e., nuclear N proteins and glycoprotein components Gc and Gn) measured against nonirradiated samples. 19 We also use transmission electron microscopy (TEM) to show that the UV treatment leaves virions physically intact with an average diameter of 193 nm. 16
Fluorescent Labeling and Quantifying of SNVR18
Hantaviruses consist of a nucleocapsid surrounded by a lipid bilayer membrane derived from membrane lipids of the host cells. As such, SNV is amenable to staining by fluorescent derivatives of lipophilic lipid probes such as octadecyl rhodamine (R18). 16 We use a combination of fluorescence and absorption measurements to quantify purified and fluorescently labeled particles, SNVR18. On average, our preparations of labeled virions are usually 500 µL stocks containing 108SNVR18/µL, each bearing ≈1 × 104 R18 probes/SNV. We typically establish a signal dynamic range and variation (Z′ score) for each batch of labeled SNVR18 by titrating SNVR18 particles to 104 cells in 10 µL volumes in a plate assay ( Fig. 1 ).
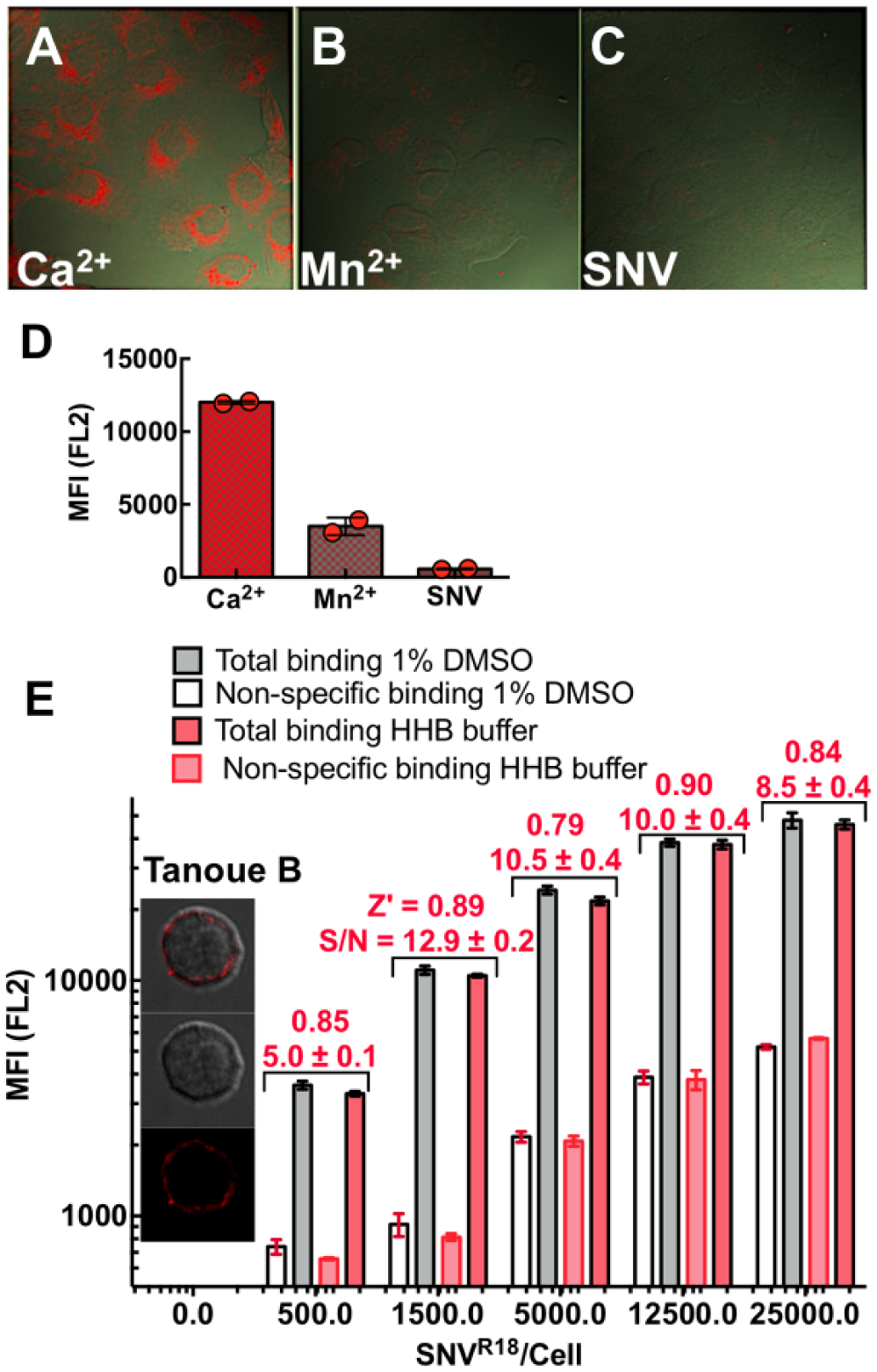
Confocal microscopy images show differential binding of SNVR18 to Vero E6 cells in HHB/0.1% HSA buffer in the presence of (
Virus Probe Validation
The median fluorescence for each well was plotted versus plate position. Before engaging in the HT screen of the PCL, we tested the suitability of our virus preparations in a pilot assay measuring reproducibility as previously described. 16 We tested the virus for specific binding to Vero E6 cells and Tanoue B cells using confocal microscopy imaging and flow cytometry. For Vero E6 cells, we used Mn2+ to block specific binding of SNV to low-affinity integrins. 16 Using a plate-based assay, we assessed signal-to-background levels using increasing concentrations of fluorescently labeled SNV (SNVR18) to a fixed number of cells, while using excess unlabeled SNV as a blocking agent. We assessed the dynamic signal range and data variation associated with these measurements concerning Z′ factors. 20
Negative controls are neat SNVR18, and positive controls are samples preblocked with 10× unlabeled SNV. An assay with Z′ scores ranging from 0.5 to 1.0 is considered excellent, whereas a Z′ score of 0 reflects a yes/no assay.
HTS Assay for Specific SNVR18/DAF Interactions in Tanoue B Cells
We performed HTS of the 1200-compound PCL distributed in four 384-well plates. A total volume of 5 µL of Tanoue B cells at a concentration of 2 × 106 cells/mL in HHB buffer was distributed to each well of a 384-well polystyrene plate, except columns 23 and 24.
HHB buffer (10 µL) was added to columns 23 and 24 for washes and to aid in well identification for analysis. In column 2, unlabeled SNV (10× concentration excess) was added as a positive control for inhibition of SNVR18 binding (2.5 µL/well). The library compounds (10 µM final concentration) were transferred to columns 3–22 using a 100 nL pin tool. The four plates were incubated at room temperature for 30 min. The SNVR18 was then added to columns 1 and 3–22 at 3 × 107 SNVR18/well in 5 µL (1.5 × 1010 SNVR18/mL), which is a 3000-fold excess of virus compared with cells. This virus-to-cell ratio was optimal for the signal-to-noise ratio (S/N; Fig. 1E ). The plate was returned to the vortex mixer for an additional 25 min at 1000 rpm. After 25 min, the plate was sampled and samples were delivered into an Accuri flow cytometer with HyperCyt automation and analyzed using HyperView software for automated well identification and cell analysis. The median fluorescence value from wells in columns 1 and 2 was used to calculate a Z′ factor.
Confirmatory Dose Response
The selected hit compounds were screened in a 10-point dose–response assay (0.1–20 µM) in the same way as the library screen. Each well, including the controls, had a constant 1% DMSO concentration, which maintained continuous light scatter characteristics of cells. The EC50 values were all in the 10 µM range.
Cell Viability Assays
We tested the hit compounds for toxicity using the CellTiter Glo kit from Promega (Madison, WI). This package assesses the level of ATP found in viable cells. Cells were subjected to a titrated concentration range of 0.1–20 µM of hit compounds over 24 h. Toxicity ≥50% was used to exclude the hit from further advancement. Toxicity was used as a factor in triaging compounds in the context of their relationship to their IC50.
Infection Assays
We assessed the ability of the compounds to prevent entry of live hantaviruses (SNV and HTNV) into Vero E6 cells in a BSL3 laboratory setting. This assay uses a quantitative real-time (qRT)–PCR readout based on TaqMan technology, which we have used extensively.19,21 It has previously been optimized for SNV, ANDV, and HTNV using a multiplicity of infection (MOI) of 0.01 and a readout 48 h after infection. Primers and probes for viral S segment have already been developed and evaluated for each virus.19,21 We plated Vero E6 cells (5 × 104/well) in a 48-well plate, and the following day, we infected separate wells in triplicate with each of the two viruses in the presence or absence of 10 µM concentrations of the HTS screen hit compound. After a 48 h incubation, we prepared supernatant RNA using viral RNA preparation kits (Qiagen, Venlo, Netherlands) and subjected the RNA to TaqMan analysis as previously described.19,21
Alternatively, infectivity is analyzed by standard Western blot analysis of N protein expression. 8 Briefly, for Western blot, SNV N protein was detected with αSNV/N (hyperimmune rabbit anti-SN virus N protein) used at a 1:1000 dilution with a secondary antibody (Peroxidase AffiniPure Goat Anti-Rabbit IgG; used at 1:1000 dilution) from Jackson ImmunoResearch Laboratories (West Grove, PA; cat. no. 111-035-003, lot no. 104668). We treated the nitrocellulose membrane with a horseradish peroxidase (HRP) substrate from Pierce, SuperSignal West Pico (Waltham, MA; product no. 0034077) before imaging. Quantitative analysis of the gel was performed using a BioRad Molecular Imager, ChemiDoc XRS+, equipped with Image Lab Software 4.1. We verified equal loading by detecting calnexin with anticalnexin monoclonal antibodies (mABs; clone H-70 used at 1:500 dilution; Santa Cruz, Santa Cruz, CA) or β-actin on the same membrane with the anti-β-actin (clone AC-74 used at 1:2000 from Sigma).
Pertussis Toxin
A total of 150,000 CHO-A24 cells were plated onto a 48-well plate for 24 h, treated with 100 ng/mL pertussis toxin (PTX), incubated for ~18 h at 37 °C, and then used in infection or GTPase activity assays.
GTPase Effector Trap Flow Cytometry Assay (G-Trap)
This assay has been previously described.8,22 PTX-treated and mock-treated CHO-A24 cells were serum starved overnight, in a 48-well plate with 50,000 cells in each well. After incubating the cells with SNVR18 for 5 min in 100 µL of media at 37 °C, we lysed the cells with 100 µL ice-cold RIPA buffer. Lysates were kept cold at all times to limit hydrolysis of active GTPases. For each sample, 50 µL was used to analyze for GTP loading of Rap1. A total of 10,000 beads functionalized with the Rap1 effector protein RAL GDS were used to capture Rap1GTP from the cell lysate as previously described.8,22
Cyclic AMP Assay
We used an Upward (U0200G) cADDis BacMam fluorescent biosensor kit purchased from Montana Molecular (Bozeman, MT; www.montanamolecular.com) 17 to assay cyclic AMP production in CHO-A24 cells after exposure to SNV. The sensor application involves a two-step process supplied by the manufacturer. It consists of inoculating cells plated in a 96-well plate with a baculovirus transduction system in appropriate media. The assay can be tested after 2 days. We optimized the assay for our system by titrating the sensor expression and measuring the response of the positive control (e.g., forskolin) relative to our SNV assay. It is essential to perform the optimization process regarding the corresponding reaction between the positive control and the test of interest (e.g., SNV). This is because excess sensor expression, which is optimal for the positive control, might be undesirable for the assay target as it limits response compared with low-expressing effectors, as in the case of SNV. We plated BacMam-transduced cells in a 48-well plate (48,000 cells/well) and tested the cells for SNV-induced cyclic adenosine monophosphate (cAMP) production using a BioTek Synergy two-plate reader (Winooski, VT). Cell activity inhibitors were added to target wells and allowed to incubate for 30 min. We recorded baseline fluorescence readings of normal and transformed cells for 5 min before adding SNV and forskolin. We performed triplicate readings for each condition. We measured the cellular response as change in fluorescence pre- and postactivation.
PAC-1 Binding Assay
We incubated CHO-A24 cells (1000 cells/µL) in HHB buffer containing 1:20 dilution PAC-1, a monoclonal IgM antibody that selectively binds to physiologically activated integrin αIIbβ3. 23 To test candidate compounds for inhibition of integrin activation, we added 10 µM aliquots of each compound to 20 µL volumes of the CHO-24 cell suspensions for 30 min. Negative control samples were treated with 1% DMSO. We then added SNV aliquots (5000 SNVR18/cell) to the cell samples maintained at 37 °C for 5 min and quenched cell activation with ice-cold buffer. The samples were incubated for 30 min to enable PAC-1 binding to active conformation integrins to reach equilibrium. The samples were centrifuged once and resuspended in 50 µL of HHB buffer and then read on an Accuri C6 flow cytometer.
Results
Assay Validation
To identify compounds that inhibit binding of SNVR18 in an HTS campaign, we first established the specificity 16 and affinity of binding of SNVR18 to DAF functionalized beads and Tanoue B cells. 12 Figure 1 shows SNVR18 binding to Vero E6 cells expressing αvβ3 integrin and DAF. SNV binds to the PSI domain of low-affinity integrins. 6 Thus, treatment of cells with Mn2+cations, which induces an extended conformation, blocks SNV binding.6,16 Here we show that treatment of cells with 1.5 mM Mn2+ inhibits SNVR18 binding to Vero E6 cells by ≥70% ( Fig. 1B ). We also show that unlabeled SNV efficiently inhibits SNVR18 from binding to Vero E6 cells ( Fig. 1D ). Because the library compounds are solubilized in DMSO, which is cytotoxic above 1% concentration by volume, we tested whether 1% DMSO had adverse effects on our assay in a simulated HTS setting. In a 384-well plate, titration of SNVR18 particles to DAF-expressing Tanoue B cells yielded comparable Z′ scores of 0.8–0.9 across a 50-fold range of virus particle titers in standard cell media and 1% DMSO buffer. Although nonspecific binding appeared to increase with increasing titers of SNVR18, the signal-to-background levels (S/N in Fig. 1E ) increased from 5 at the lowest SNVR18 titer to ~13, before decreasing to ~9-fold at the highest titer of SNVR18.
Primary HTS
The median fluorescence for each well was plotted versus plate position ( Fig. 2A ). The average Z′ score for the plates was ~0.5, indicating that the assay had appropriate statistical reliability for HTS. As shown, the library compounds comprising fluorescent, light-emitting compounds, and compounds that quenched SNVR18 fluorescence emission below the positive control were not considered. We calculated the percent inhibition based on the following equation: % Inhibition = 100[1 – ((MFItest – MFIblocked)/(MFIunblocked – MFIblocked))], where MFIi is the median fluorescence intensity of cells in wells containing test compounds, blocked control wells, and unblocked controls, respectively. From these screens, 24 compounds were hits, inhibiting the binding response of the virus to the Tanoue B cells at >70%. We also analyzed forward and side scattergrams (FSC and SSC in Fig. 2B ) for clear indications of cytotoxicity. Dead or dying cells are expected to become smaller (reduced FSC) or more granular (increased SSC). In general, the scattergrams for the vehicle (DMSO-only) treated cells and noncytotoxic compounds displayed ≥80% events in the gate associated with healthy cells. We observed an increase in log SSC and a decrease in the percentage of the population of viable cells, resulting from treatment with cytotoxic compounds (e.g., 73% viable cells treated with fenbendazole in Fig. 2B ).
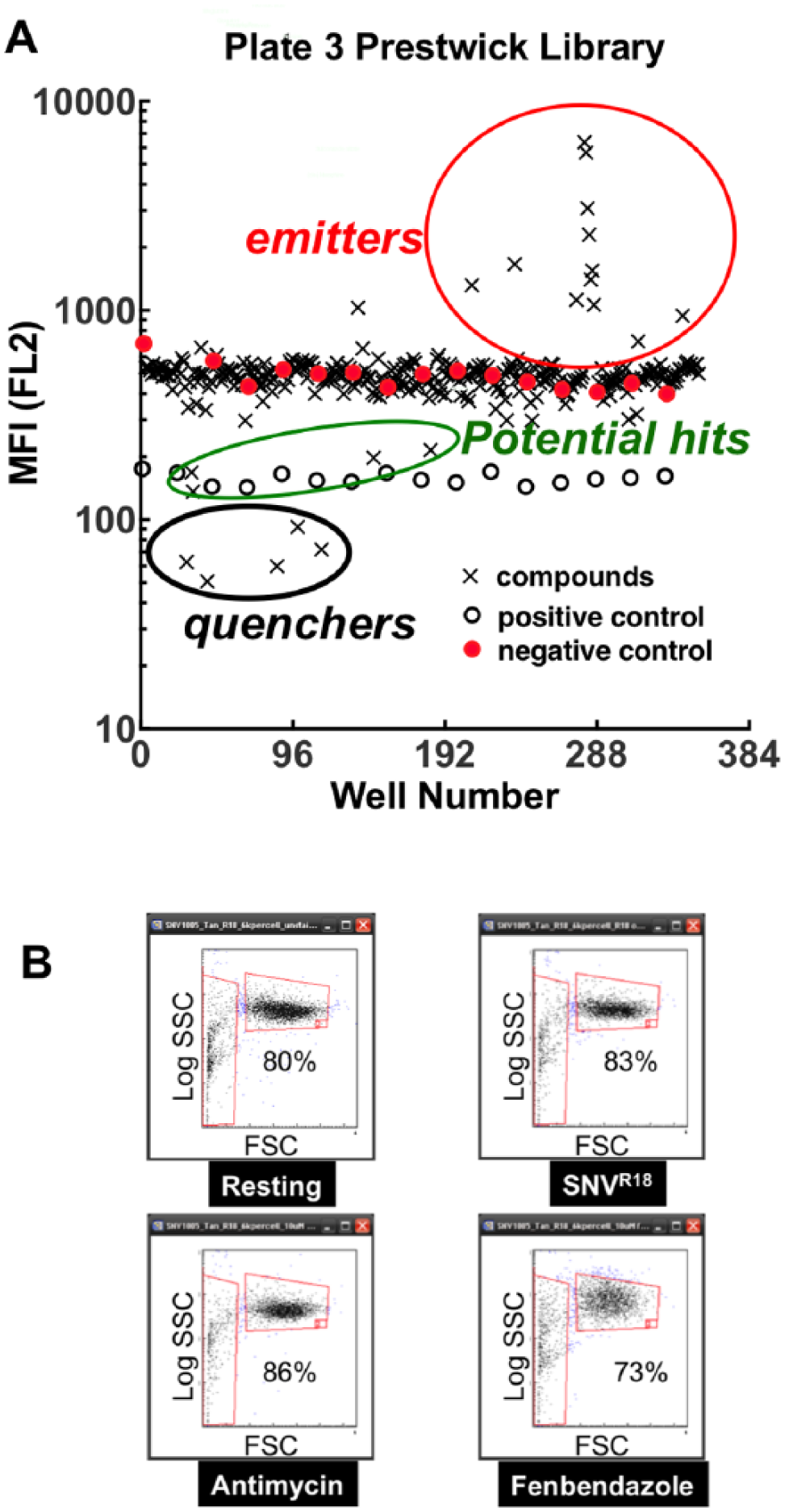
HTS of the PCL. (
We performed dose–response measurements by testing the effects of compounds at 10 concentrations ranging in a half-log dilution series from the nanomolar to the micromolar range. Eight compounds were confirmed (gossypol, trifluoperazine, niclosamide, nifedipine, tocopherol, fenbendazole, antimycin, and tolfenamic acid) by showing reduced SNVR18 fluorescence in a dose-dependent manner. We fitted the data to a dose–response inhibition curve with a variable slope in GraphPad Prism Software Version 6.0h. IC50 values ranged from 0.5 to 40 µM ( Suppl. Fig. S1 ).
Confirming the Activity of Hit Compounds
We next tested the candidate compounds for infection inhibition and cytotoxicity. Vero E6 cells were incubated with the eight candidate compounds for 30 min before infection with 0.1 MOI SNV and HTN inocula. Results indicated that three compounds inhibited the infection of Vero E6 cells by SNV and HTN: niclosamide (~60% SNV, ~70% HTN inhibition), gossypol (~70% SNV and HTN), and antimycin (≥80% SNV and HTN) ( Fig. 3A ). However, the five other compounds failed to inhibit infection significantly. Four of the compounds inhibited infection by ≤20%, whereas fenbendazole inhibited infection by nearly 40%, which is below our set threshold for acceptability in our primary screen. We then subjected Vero E6 cells to doses of compounds spanning a concentration range of 0.1–20 µM over 24 h in a 96-well plate, to test cells for viability after long-term exposure to hit compounds. We determined toxicity using the CellTiter Glo kit from Promega. Figure 3B shows the concentration-dependent effects of the eight compounds on cell viability. Niclosamide (~50% viability) and gossypol (~30% viability) were notably cytotoxic. Fenbendazole (~70%) was moderately cytotoxic with increasing concentration of the drugs. Antimycin, nifedipine, and tocopherol had minimal cytopathic effects after 24 h at the limiting concentration of the HTS assay.
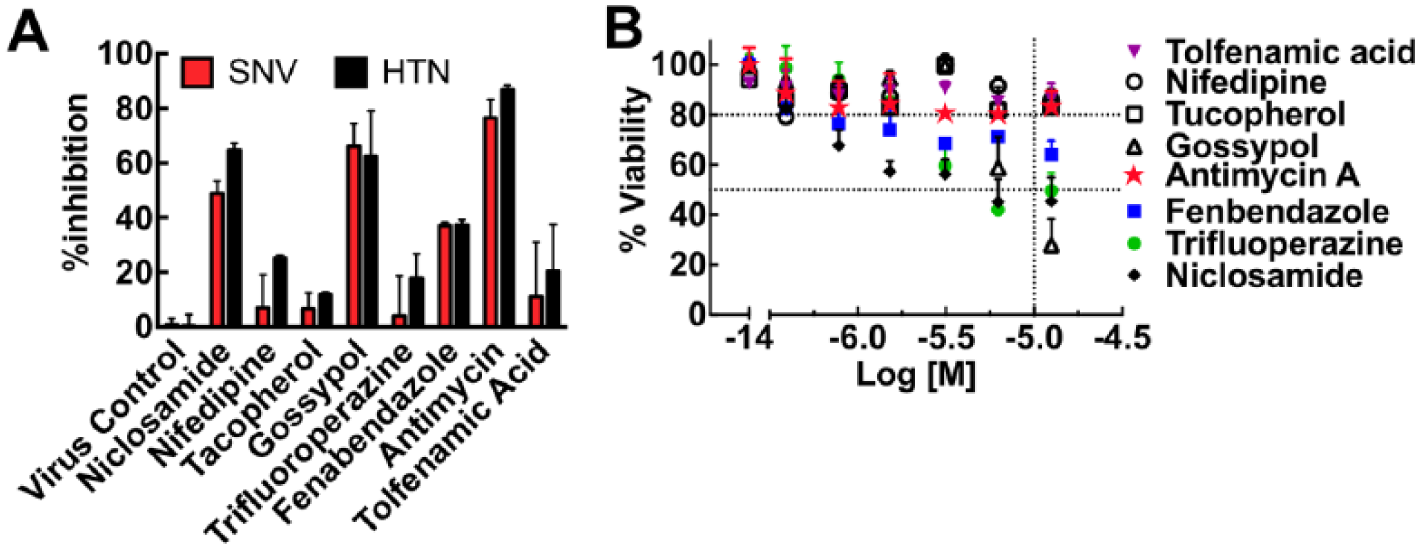
Cytotoxic hits yield false-positive results on hantavirus infectivity. (
Distinction of Fluorescence Quenching from Competitive Virus Displacement
Having identified antimycin as an active inhibitor of infection with minimal direct cytotoxicity, we performed additional competitive binding experiments to investigate its mechanism of action. We tested the compounds for probe quenching and determined that the compound characteristics were consistent with molecules that formed nonfluorescent complexes or aggregates upon interacting with R18. The quenching mechanism is commonly described as static quenching 24 ( Suppl. Fig. S2 ). Thus, a decrease of cell-associated fluorescence in our screening assay could feasibly have resulted from quenching of SNV-bound R18 rather than (or in addition to) competitive displacement of the virus by the test compound, a false-positive result. Quenching can be distinguished from competitive displacement by the kinetics with which fluorescence is diminished. We therefore used soluble DAF (sDAF) to compete off the SNVR18 from DAF functionalized beads as a positive control for the kinetics of competition binding. Over a 3 min time course of analysis on a BD FACScan flow cytometer, antimycin and two other of the hit compounds (gossypol and tolfenamic acid) promoted a precipitous drop (koff ~ 0.25 s−1) in bead fluorescence ( Fig. 4A,B ). By contrast, beads treated with 10 µM sDAF displayed a single exponential decay curve of SNVR18 (koff ~ 0.01 s−1; Fig. 4B ). The significant discrepancies in fluorescence loss kinetics suggested that static quenching confounded the competition binding assays.
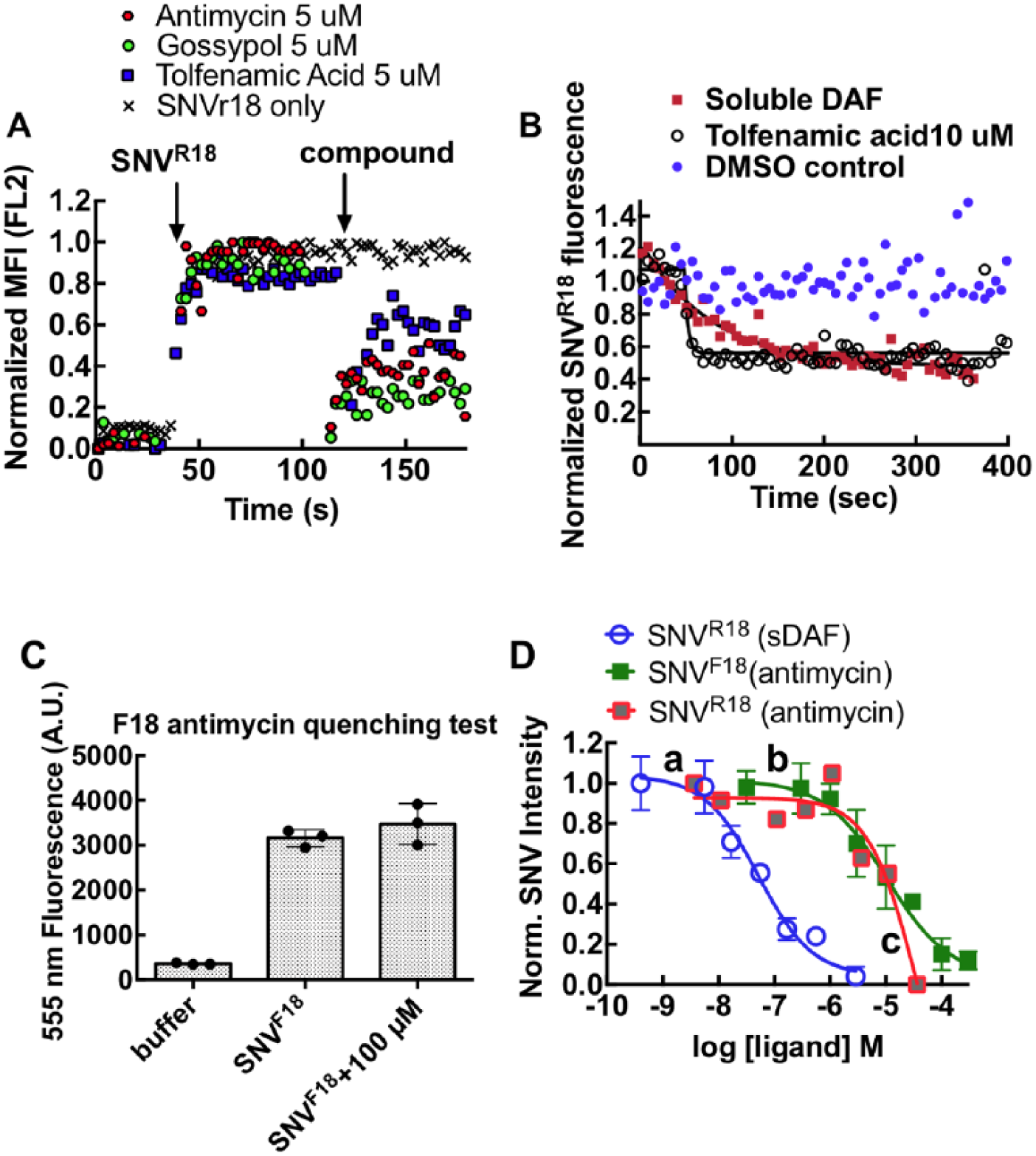
Investigation of fluorescence reporter interference by candidate hit compounds. (
Electrostatic attraction and aromatic stacking interactions are often associated with static quenching. 24 R18 is a positively charged fluorophore; we therefore tested whether SNV particles labeled with the negatively charged F18 were refractory to quenching by antimycin. We first established that antimycin does not quench SNVF18 fluorescence by mixing antimycin with SNVF18 ( Fig. 4C ). We then compared the dose–response curves for antimycin in binding competition with SNVR18 and SNVF18 with Tanoue B cells using a flow cytometer ( Fig. 4D ). We used sDAF titrations to compete with SNVR18 in a binding assay to DAF functionalized beads, as a standard control for competition binding. 12 The data were fit to a competition binding curve (Y = Bottom + (Top – Bottom)/(1 + 10^((LogIC50 – [Ligand])*Hill slope))) using GraphPad Prism 6.0 h. None of the parameters were fixed, and the unfixed hill slopes were not significantly different (~ –0.8), indicating single site binding. Fixing the hill slopes to unity did not significantly change the fit parameters. The SNVF18 antimycin curve was characteristically similar to the sDAF curve but shifted to the right due to lower affinity for antimycin (IC50 = 50 nM for sDAF vs 1C50 = 10 µM for antimycin). Normal competition curves characteristically show that the dose–response concentration ranges of the target ligands span more than two orders of magnitude, 25 as indicated for sDAF versus SNV R18 and antimycin versus SNVF18. Conversely, the effective concentration range of antimycin titrations used in the antimycin–SNVR18 curve was relatively narrow, suggesting that the process did not represent an actual competition binding curve, but one confounded by probe quenching.
SNV Binding to Low-Affinity Integrin Stimulates Cyclic AMP Production
We have recently shown that SNV engagement of the integrin PSI domain involves the transduction of tensile force transmitted through the integrin. 8 Others have shown that force-stimulated integrins might recruit stress-dependent signaling molecules to focal adhesions, including cyclic AMP, by activating Gαs at the site of force application. 26 For our purposes, Gαs stimulation results in integrin activation, by a pathway involving cAMP and the exchange protein activated by cAMP (Epac) and Rap1. 27 As a rationale for establishing a cyclic AMP assay for assessing the mechanism of action of our inhibitor compounds, we first tested whether SNV binding to the integrin PSI domain activated the Gαi/Gαs signaling axis ( Fig. 5A ). 28 These assays used DAF-null CHO-A24 cells, stably expressing integrins αIIbβ3 and P2Y2R receptors. 8 We then used a Gαs-selective antagonist NF449 29 to determine whether Gαs signaling was necessary for SNV infection of CHO-A24 cells. We used a Western blot to measure SNV N protein expression as described in the methods. We established loading controls of the gels by using a typical standard housekeeping gene, such as β-actin, on the same membrane. Quantitative analysis of the gel was performed using a BioRad Molecular Imager (Hercules, CA). NF449 reduced the viral N protein by >80% relative to vehicle-treated cells, indicating that Gαs signaling was essential for infection ( Fig. 5B ). After that, we tested whether Gαi signaling was involved in SNV infectivity by using PTX to block Gαi activity. Inhibition of Gαi signaling with PTX increased viral N protein twofold relative to control cells ( Fig. 5C ). As a matter, of course, we then compared GTP loading of Rap1 in PTX- and non-PTX-treated cells 5 min after exposure to UV-killed SNVR18. The nearly twofold increase in Rap1-GTP in PTX-treated cells was comparable to the rise in infection ( Fig. 5D,E ), indirectly showing that cAMP stimulation was a decisive factor in the infection of CHO-A24.
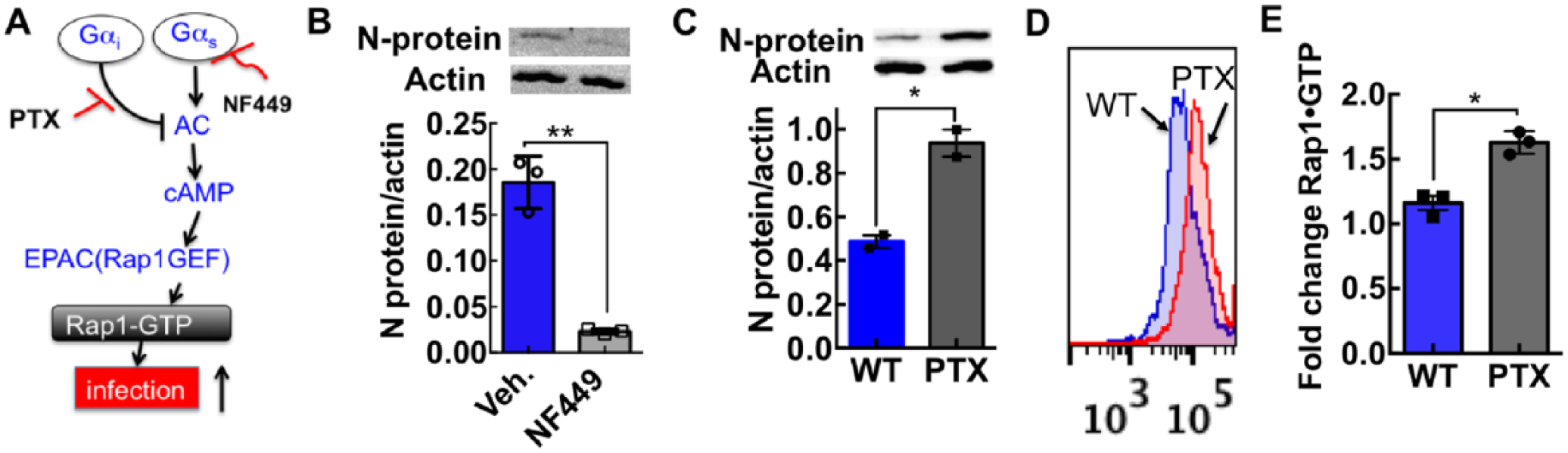
Cyclic AMP stimulation potentiates SNV infectivity. (
Antimycin Inhibits cAMP Stimulation by Directly Binding to SNV
We measured SNV-induced cAMP production in 48,000 CADDis-transformed CHO-A24 cells under various conditions ( Fig. 6A ). As shown, SNV binding to low-affinity β3 integrins stimulated cAMP signaling (red squares). Preincubating antimycin with SNVR18 and adding to the cells (100 µM antimycin + SNV in Fig. 6A ) inhibited stimulation of cAMP more effectively than incubating antimycin with cells before the addition of SNVR18 ( Fig. 6B ; also see Suppl. Fig. S3 for kinetic data). This result suggested that antimycin binds directly to the virus and not to the cell receptor. We treated other wells with 1.5 mM Mn2+ and 10 µM GRGDSP, followed by SNV. We used Mn2+ as a positive control for inhibition of SNV binding to integrins.6,16 Also, Mn2+ was applied to determine whether Mn2+-induced integrin activation caused cAMP production. The data show that Mn2+, which prevented SNV binding to the cells,6,16 did not affect cAMP production. We used the soluble fibronectin hexapeptide GRGDSP to disrupt the putative cis interaction of αIIbβ3 with RGDP2Y2R in CHO-A24 cells. Interestingly, GRGDSP did not affect SNV-stimulated cAMP response, indicating that cAMP stimulation is not integrin binding to an anchored ligand.
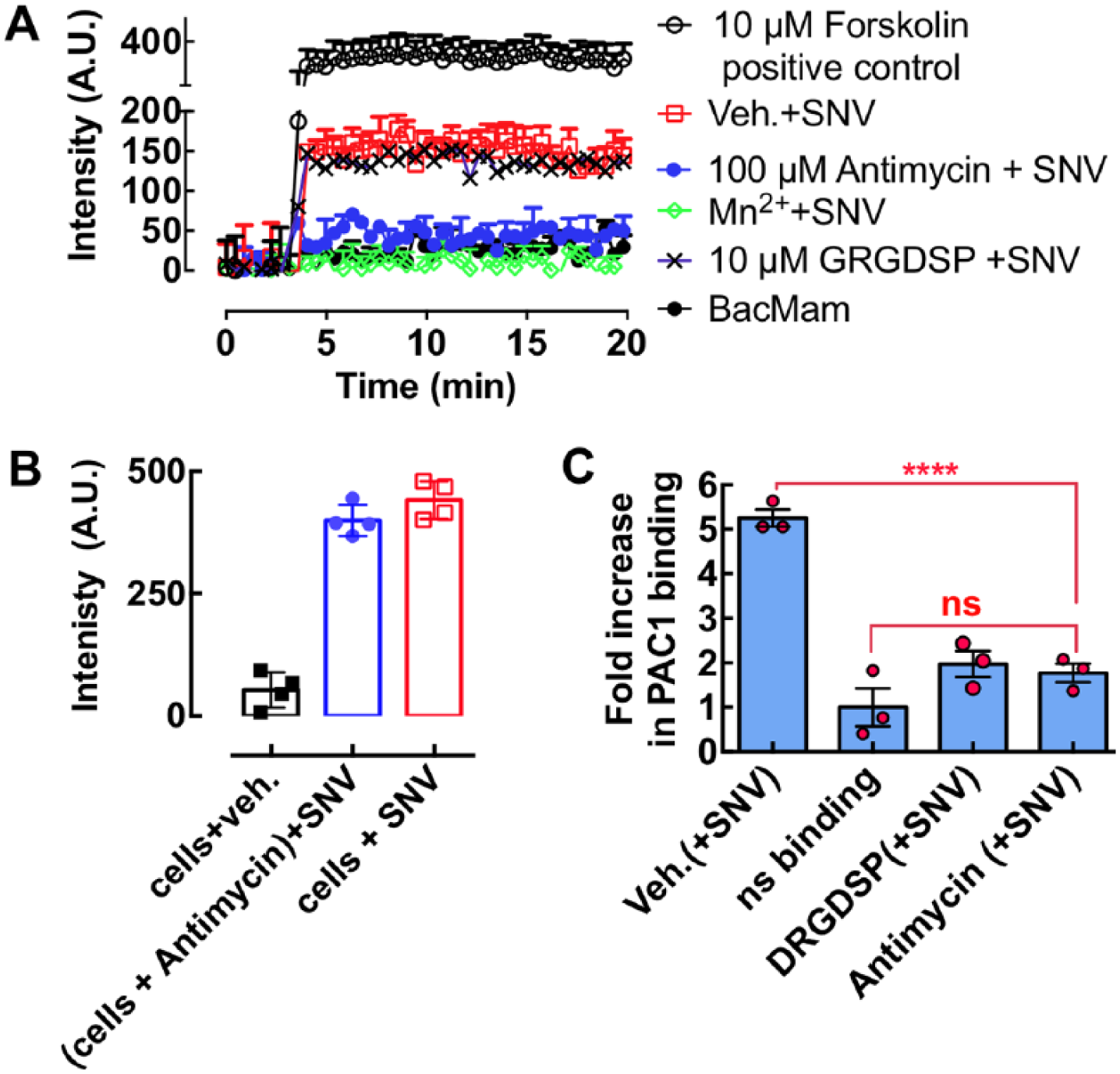
(
Testing Antimycin for Inhibition of Full αIIbβ3 Integrin Activation
SNV binds to the PSI domain of low-affinity integrins interacting in cis with P2Y2R and stimulates outside-in signaling. Outside-in signaling requires a surface-anchored ligand, such as P2Y2R, which can support traction force on its exertion.8,9 The resulting changes in conformation to full activation allow the integrin’s ligand binding site to be recognized by PAC-1, an integrin activation-dependent IgM antibody. 8 Exposure of CHO-A24 cells to SNV stimulated a fivefold increase in PAC-1 staining above nonspecific background (Veh. and ns binding in Fig. 6B ). Antimycin preincubated with SNV inhibited PAC-1 staining (antimycin in Fig. 6B ), whereas preincubating antimycin with cells produced irreproducible results (data not shown). GRGDSP (GRGDSP [+ SNV] in Fig. 6B ) inhibited PAC-1 staining, unlike cAMP ( Fig. 6A ). Thus, this result reveals that cAMP stimulation is an upstream signaling event relative to signaling associated with the integrin conformational change required for the molecular recognition of PAC-1. 8
Discussion
We demonstrate that a BSL3 pathogen, SNV, can be produced and inactivated in sufficient quantities of known concentration12,16 for use in an HTS campaign. We have performed HTS of the 1200-compound PCL with a homogeneous flow cytometry-based assay to identify compounds that can inhibit SNV binding to DAF. We have identified one compound, antimycin. Antimycin blocked binding of SNV to integrin β3-null but DAF-replete Tanoue B cells. Antimycin inhibited infection of Vero cells that express DAF and integrin β3. Antimycin inhibits integrin-associated cAMP production and molecular recognition by PAC-1, an IgM antibody that binds to activated integrins. Therefore, the results suggest that antimycin’s molecular mechanism of action is interacting with the hantavirus’s receptor binding Gn/Gc proteins, which in turn interferes with the virus’s ability to bind cellular receptors.
Antimycin is a specific inhibitor of respiration and is the active component found in the commercial fish poison Fintrol. The value of antimycin as a candidate compound is based on the known, structural details of its interaction with counterstructures associated with its bioactivity.30,31 Thus, such information could be used in structure–activity relationship optimization as well as identification of structural homologs, which might serve as templates for optimization. Our secondary receptor-blocking assays are carried out in a short time frame that rules out the possibility of cytopathic effects.
It is worth noting that antimycin shares common features with small antiviral compounds that inhibit infection by partitioning into the envelope membranes of diverse viruses such as influenza and hepatitis C. These compounds block infectivity of unrelated viruses by inhibiting fusion of the viral envelope membrane to the host cell.32,33 While antimycin associates with the viral envelope and thus quenches R18, like the other hits found in our study, antimycin blocks SNV binding to cells, which is in contrast to the other HTS hit compounds that quenched R18 but did not inhibit infection.
The most significant aspect of our study is the development of secondary homogenous cell suspension assays to screen for small-molecule inhibitors of SNV-induced integrin activation. These tests are compatible with flow cytometry and the 96- or 384-well plate format, and therefore amenable to HTS configuration. Integrin activation plays a critical role in many physiological processes, including activation of the immune response, inflammation, hemostasis, thrombosis, cell migration, and tumor cell invasion. Thus, discovery campaigns to understand the signaling pathways and their modulation are important steps in controlling the diseases associated with dysregulated integrin activity.34–36
It is important to note that the concept of SNV-induced integrin activation postoccupancy of the PSI domain of low-affinity integrins is novel and has only been recently described. 8 The canonical switchblade model of physiological integrin activation postulates that resting integrins assume a low-affinity bent conformation for their ligand and upon full activation, integrins acquire an extended structure with an open headpiece. 37 Integrin αIIbβ3 thus activated by SNV recognizes the PAC-1 antibody. 8 The intermediate signaling steps that occur between SNV engagement and molecular recognition of PAC-1 by the fully active integrin have not been explored. In this study, we show that cAMP production is an early downstream consequence of SNV engagement. cAMP is usually activated downstream of ligand-occupied heterotrimeric GPCRs that associate with the Gαs subunit. In this setting, active Gαs (GTP bound) can induce the plasma membrane–bound adenylyl cyclase to catalyze the conversion of ATP to cAMP. 38 Direct binding of cAMP to Rap1–guanine nucleotide exchange factor (Rap1-GEF) Epac1 provides a Gαs-dependent mechanism for SNV-induced physiologic integrin activation.39,40 Our study relies on inhibition assays of Gαi and Gαs to support the idea that the stimulation of cAMP accumulation is linked to Gαs activation. 26 In sum, our study exploits two mechanically transduced signal transduction steps, cAMP accumulation and change in integrin conformation, to validate the HTS discovery target, antimycin, as a bona fide inhibitor of SNV interaction with cell entry receptors.
We propose to exploit lessons from this study to extend the original HTS screen to a two-color HTS screen. Thus, instead of using Tanoue B cells for a primary screen, we propose to use CHO-A24 cells, where SNV binding to the PSI domain of the integrin can be measured directly in one fluorescence channel (FL2) in tandem with integrin activation as reported by PAC-1 staining in an alternate fluorescence channel (FL1). Cell-associated SNV fluorescence would monitor direct inhibition of SNV binding, and modulation of PAC-1 staining could suggest a mechanism of action. High-throughput flow cytometry would be the platform of choice for this activity.
Supplemental Material
DISC766623_Supplemental_Material – Supplemental material for A High-Throughput Flow Cytometry Screen Identifies Molecules That Inhibit Hantavirus Cell Entry
Supplemental material, DISC766623_Supplemental_Material for A High-Throughput Flow Cytometry Screen Identifies Molecules That Inhibit Hantavirus Cell Entry by Tione Buranda, Catherine Gineste, Yang Wu, Virginie Bondu, Dominique Perez, Kaylin R. Lake, Bruce S. Edwards and Larry A. Sklar in SLAS Discovery
Footnotes
Supplemental material is available online with this article.
Declaration of Conflicting Interests
L. A.S and B.S.E. hold patents for HyperCyt technology and are cofounders of IntelliCyt, a company that manufactures and sells instruments incorporating HyperCyt technology. No financial support was received from any commercial entity for the production of this work.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIH grants R21NS066429 and MH084690, the UNM Department of Pathology, and the School of Medicine RAC award.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.