
Abstract
Lysophosphatidic acid (LPA) activates the G-protein-coupled receptor LPA1, which regulates various cellular processes, including cell proliferation and migration. Although LPA1 transduces cellular responses via Gq, Gi, and G12/13, associations between these signaling molecules and cellular phenotypes remain poorly characterized due to the lack of signal-specific pharmacological tools. Here, we characterized novel signal-biased modulators using multiple assays, including label-free impedance assays. LPA caused dramatic changes in cellular impedance in LPA1-expressing recombinant cells, which were susceptible to G-protein and protein kinase inhibitors. Subsequently, Gq-biased LPA1 negative allosteric modulators (NAMs) were identified using high-throughput screening, and a nonbiased antagonist differently affected the LPA-induced cellular impedance. These NAMs provide pharmacological tools for further investigations of the biology of LPA1.
Keywords
Introduction
Lysophosphatidic acid (LPA) is a bioactive lipid mediator that activates diverse cellular actions via the LPA receptors LPA1–6.1,2 LPA1 is ubiquitously expressed in human adults and has been detected in the brain, uterus, testis, lung, spleen, skeletal muscle, and other tissues. LPA1 is involved in various cellular functions, such as cell proliferation, adhesion, migration, survival, and cell–cell interactions.1,2 Recent studies have revealed the relevance of LPA1 function to multiple human disorders, including neuropathic pain, 3 fetal hydrocephalus, 4 cancer,1,5 and fibrosis. 6 Currently, the LPA1 antagonists BMS-986020 and SAR100842 are in clinical development for the treatment of idiopathic pulmonary fibrosis, systemic sclerosis, and related fibrotic diseases. 7
LPA1 is a G-protein-coupled receptor (GPCR) that couples with the G-proteins Gq, Gi, and G12/13 to induce downstream cellular responses and recruit β-arrestin upon receptor activation.1,2,7,8 Although these signals have varied effects on cellular functions, the details have not been fully elucidated because of their molecular complexity. Cellular phenotypes that are regulated by LPA are dependent not only on these complicated signals, but also on cell types. For example, LPA reportedly promotes the migration of vascular smooth muscle cells through the activation of Gq, which is important for the development of intimal hyperplasia after vascular injury,9,10 whereas G12/13- and Gi-mediated signals contribute to LPA-stimulated cell migration in ovarian cells.11–13 Because these conclusions are based on studies using G-protein inhibitors and relevant signal inhibitors that do not target LPA1, it remains unclear whether signal-specific modulation of LPA1 results in the same phenotype.
Recently, ligands that preferentially activate one or more intracellular signals, a phenomenon referred to as “signaling bias” or “functional selectivity,” have been attracting considerable interest because they may provide new possibilities for developing drugs with more therapeutically beneficial profiles.14,15 For example, the G-protein-biased opioid receptor agonist TRV130 is in development as an alternative analgesic to morphine that may avoid gastrointestinal dysfunction and respiratory depression.14,16
Traditionally, the actions of drugs on GPCRs have been assessed using multiple assays. However, the complete description of biased ligands is often challenging because the approaches toward it are based on monitoring a single signaling event, often at a single time point. Under these circumstances, emergent label-free technologies have recently facilitated the identification of signaling biases and classification of biased ligands.17,18 Among these technologies, kinetic measurements of cellular impedance reflect changes in global cellular features, including adhesion and morphology regulated by intracellular signal transductions. This technology has been used to monitor cellular signaling processes mediated by a number of GPCRs.19–23 In some cases, impedance assays also facilitate the detection of unexpected characteristics/features of test compounds. 24
Here, we identified novel Gq-biased LPA1 negative allosteric modulators (NAMs) that distinctly modulate LPA-induced cellular impedance from nonbiased antagonists. The effects of these NAMs on cellular impedance were compared with those of signal inhibitors for G-proteins (Gq and Gi) and kinases (ROCK and GRK2), which are thought to be regulated downstream of GPCR activation. These NAMs may be useful pharmacological tools for better understanding of LPA1 function and the subsequent development of therapeutics.
Materials and Methods
Materials
Cmpd101 hydrochloride was synthesized by Takeda Pharmaceutical Company (Fujisawa, Japan). LQ1 and LQ4 were purchased from Enamine Ltd. (Kiev, Ukraine). LQ2, LQ3, and LQ5 were obtained from TimTec (Newark, DE), Cerep SA (Poitiers, France), and Pharmeks Ltd. (Moscow, Russia), respectively. Rat hepatoma McA-RH7777 cells were purchased from DS Pharma Biomedical (Osaka, Japan). Human astrocytoma 1321N1 EA-Arrestin 2 (1321N1-arrestin) cells were purchased from DiscoveRx (Fremont, CA). Fluo-4 AM and probenecid were purchased from Dojindo Laboratories (Kumamoto, Japan). Other materials were obtained from Wako Pure Chemical Industries (Osaka, Japan) unless noted otherwise.
Cell Culture
McA-RH7777 cells stably expressing human LPA1 receptors (RH7777-LPA1 cells) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin.
Ca2+ Mobilization Assay
RH7777-LPA1 cells (10,000 cells/well) were plated in black-walled, clear-bottomed, poly-
cAMP Assay
cAMP levels were measured using HTRF cAMP dynamic 2 kits (Cisbio, Codolet, France) according to the manufacturer’s instructions. RH7777-LPA1 cells (2000 cells/well) were incubated with various concentrations of test compounds, 100 nM Forskolin, and 300 nM LPA in stimulation buffer (HBSS containing 20 mM HEPES [pH 7.5], 0.1% fatty acid–free BSA, and 500 µM IBMX) for 30 min at room temperature. Subsequently, cAMP-d2 solution and anti-cAMP-cryptate Tb conjugate were added, and the plates were incubated for 1 h at room temperature in the dark. Accumulations of cAMP were measured using Envision (PerkinElmer Inc., Waltham, MA).
β-Arrestin Recruitment Assay
1321N1-arrestin cells were transfected with ProLink-tagged human LPA1 using lipofectamine LTX (Life Technologies, Carlsbad, CA) and were seeded at 10,000 cells/well in poly-
Impedance Measurements
Cellular impedance was measured using an xCELLigence system (ACEA Biosciences, San Diego, CA). Changes in the measured impedance were defined as the cell index (CI) values. RH7777-LPA1 cells (60,000 cells/well) were cultured in 96-well E-plates overnight in culture medium at 37 °C in the presence of 5% CO2. E-plates were coated with 1.7 µg/well poly-
Data Analysis
Data were analyzed using Prism 5 software (GraphPad Software Inc., San Diego, CA). IC50 values were determined by fitting the data to a sigmoidal dose–response equation.
Results
Discovery of Gq-Biased LPA1 NAMs
To identify Gq-biased LPA1 antagonists, high-throughput screening (HTS) of the approximately 800,000-compound library at 3 µΜ was conducted using Ca2+ mobilization assays in RH7777-LPA1 cells stimulated with the LPA agonist at 300 nM (~EC80). Robust assay performance was observed throughout HTS with an average Z′ factor of 0.77, and 0.72% of compounds showed >30% inhibition in the primary screen. Following primary HTS, 34 compounds were identified as hits through the counterscreens against ATP-induced Ca2+ responses (10 µM ATP, ~EC80) by endogenously expressed ATP receptors. To assess functional selectivity, these compounds were further examined in cAMP Gi assays. In these assays, the LPA1 antagonist Ki16425 inhibited LPA-induced cAMP reductions with inverse agonist activity, whereas the five compounds LQ1–5 ( Fig. 1 ) did not show any antagonistic activity at up to 30 µM ( Fig. 2A and Suppl. Fig. S2 ). These compounds were also examined in β-arrestin recruitment assays. Ki16425 potently inhibited LPA-induced β-arrestin recruitment, whereas LQ1–5 had little effect on the β-arrestin pathway ( Table 1 ). On the other hand, these five compounds inhibited LPA-induced Ca2+ responses in a dose-dependent manner, with pIC50 values of 6.59 ± 0.23, 6.79 ± 0.18, 6.50 ± 0.20, 7.78 ± 0.11, and 6.25 ± 0.18, respectively, indicating Gq-biased antagonism ( Fig. 2B and Table 1 ).
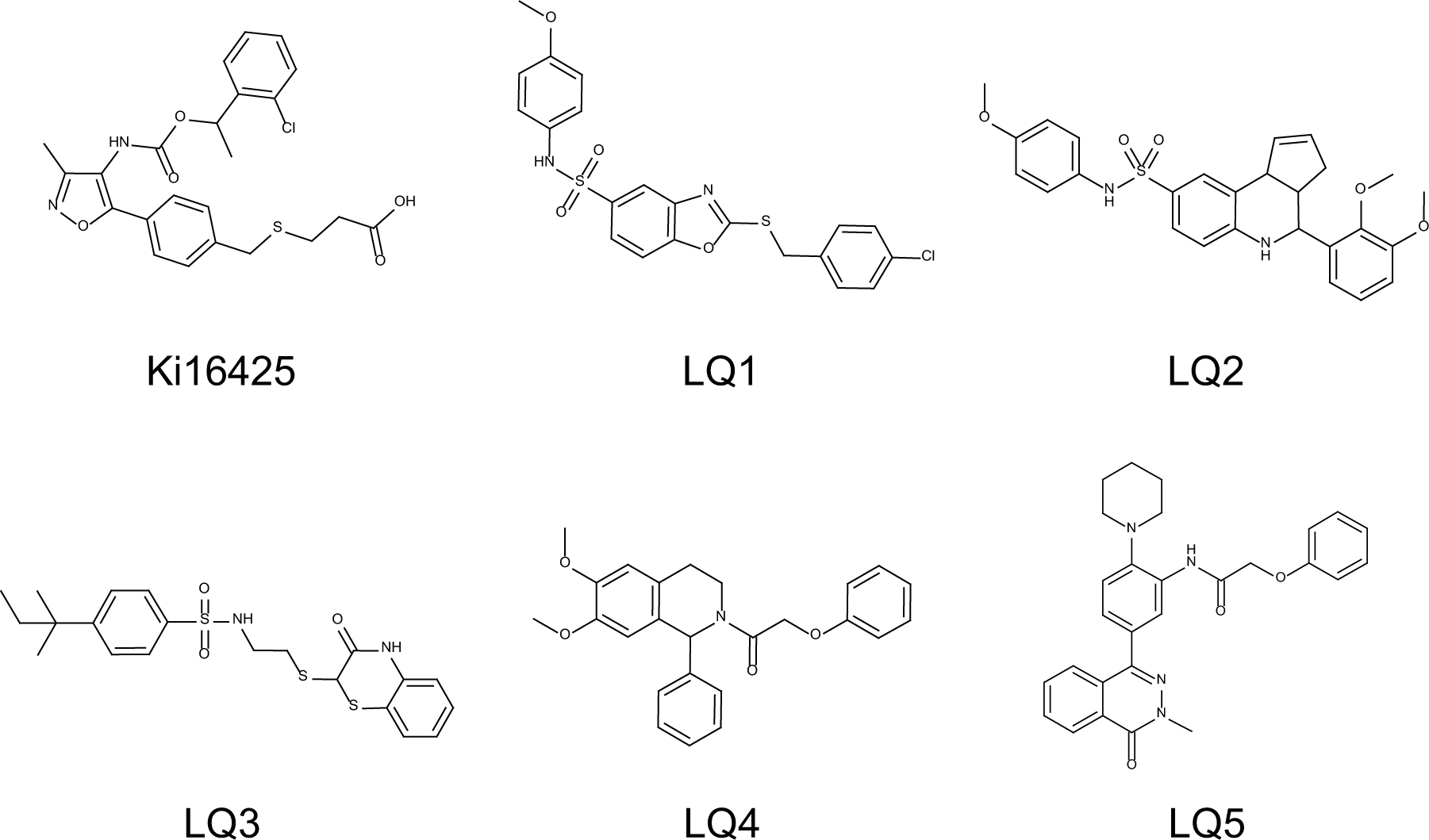
Chemical structures of LPA1 NAMs.
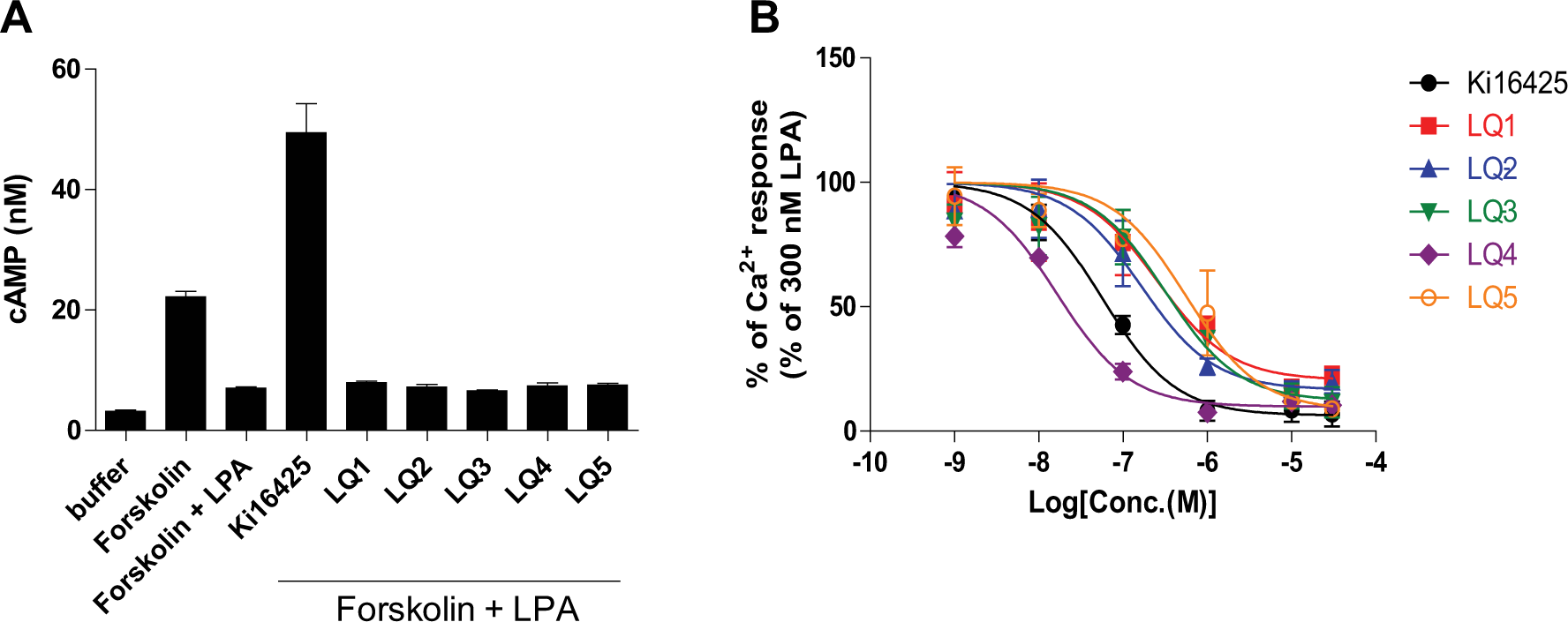
Inhibitory activities of compounds in cAMP and Ca2+ mobilization assays. (
Profiles of LPA1 NAMs.
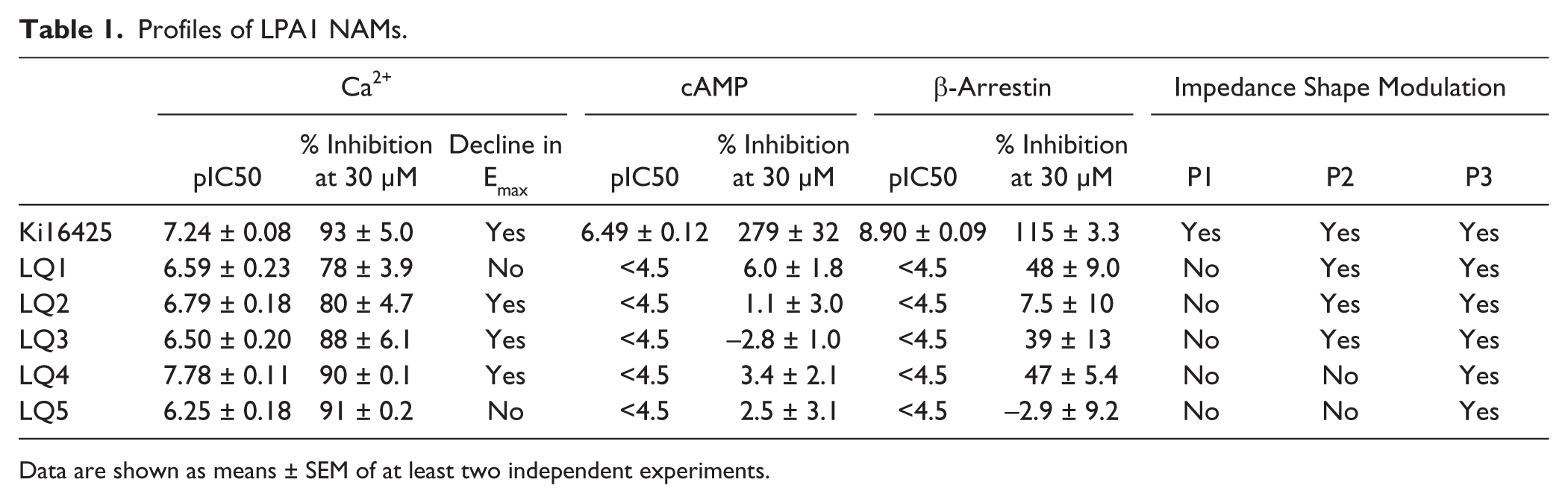
Data are shown as means ± SEM of at least two independent experiments.
To characterize the modes of action, the effects of these compounds on LPA dose–response curves (DRCs) of Ca2+ responses were examined. LQ1 and LQ5 showed saturable rightward shifts of LPA DRC, indicating that these compounds are NAMs ( Fig. 3 ). In addition, LQ2–4 and Ki16425 reduced the Emax of LPA responses, reflecting insurmountable inhibition. However, these effects were saturable at high doses ( Fig. 3 ), suggesting that these compounds are NAMs that modulate the signaling efficacy of LPA.
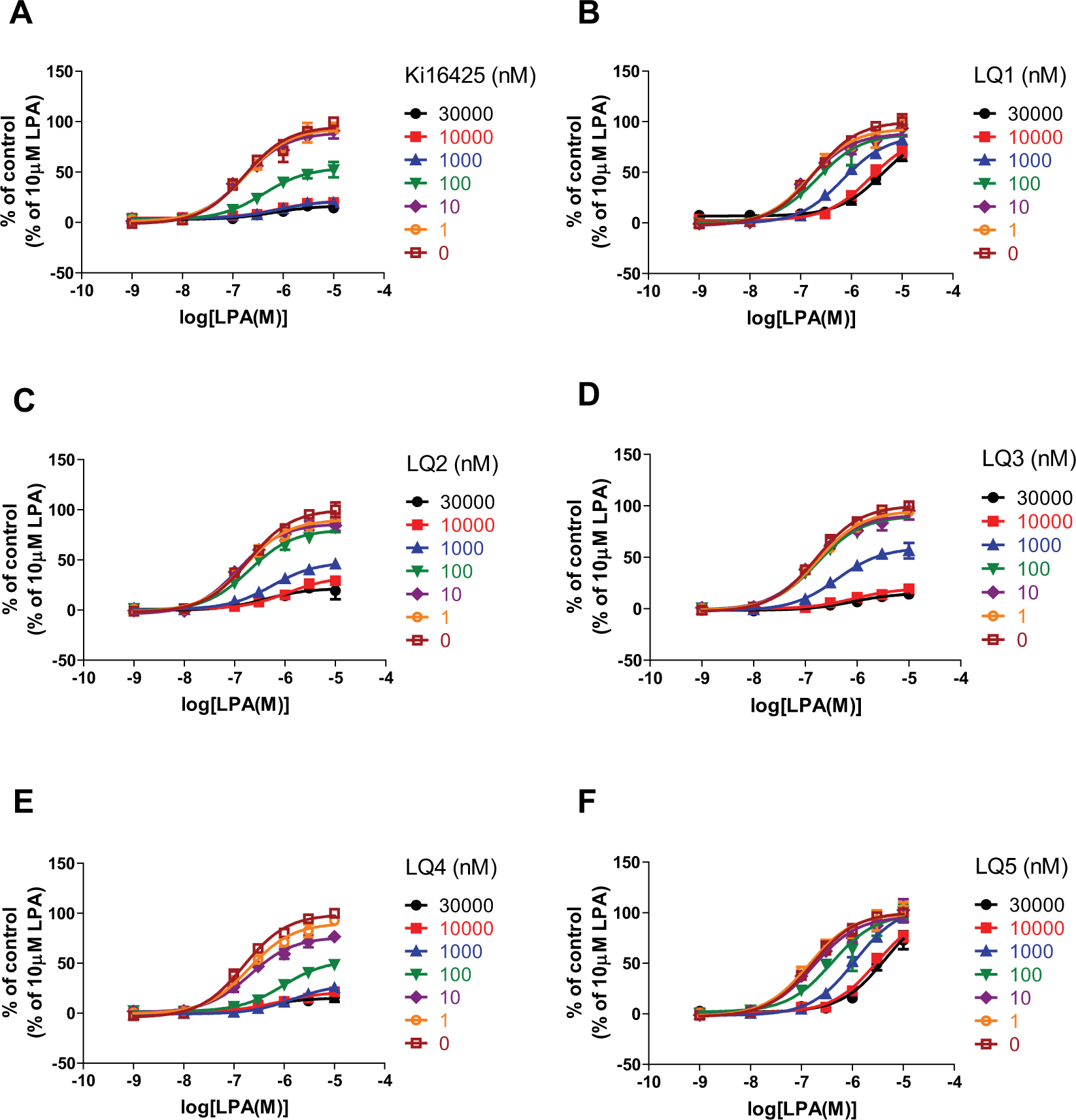
Effects of compounds on LPA DRCs in Ca2+ assays. RH7777-LPA1 cells were stimulated with a range of LPA concentrations in the presence of indicated concentrations of compounds. Data are presented as means ± SEM (n = 4).
Profile of LPA-Induced Impedance Changes
To further characterize signaling biases, LPA-mediated changes in cellular impedance were examined ( Fig. 4 ). Stimulation of RH7777-LPA1 cells with 100 nM LPA caused rapid decreases in CI (“rapid decay phase,” P1 in Fig. 4 ), followed by reverse effects (“ascending phase,” P2) and then slow decreases in CI (“slow decay phase,” P3). No responses were observed in RH7777 host cells, indicating that these responses were mediated by LPA1 ( Suppl. Fig. S3 ). To validate the intracellular signals that affect changes in cellular impedance, the effects of signal inhibitors were examined. In these experiments, the effects of G-protein and kinase inhibitors on LPA-mediated changes in impedance varied. In particular, the Gi inhibitor PTX attenuated initial decreases in CI and subsequent reverse effects. In the presence of the ROCK inhibitor Y-27632, LPA caused rapid initial increases in CI. These observations likely reflected G12/13 activation because ROCK is a downstream kinase of G12/13, which is a G-protein activated by LPA1. These results indicated that G-protein-mediated intracellular responses directly affected impedance changes. Conversely, the Gq inhibitor UBO-QIC negligibly changed the shape of LPA-induced impedance despite completely inhibiting LPA-induced Ca2+ responses ( Suppl. Fig. S4 ). These data indicated that Gq-mediated signaling was not directly involved in cellular impedance changes. We also examined the effects of the GRK2 inhibitor Cmpd10125,26 because phosphorylation of GPCRs by GRK2 leads to the recruitment of the scaffold protein β-arrestin, which is involved in not only receptor internalization but also various intracellular signaling pathways in many GPCRs. 27 Indeed, Cmpd101 inhibited LPA-induced β-arrestin recruitment with a pIC50 value of 4.99 ± 0.28 in the PathHunter assay (data not shown). In these experiments, Cmpd101 did not markedly affect the shape of the LPA-induced impedance changes and tended to enhance them ( Fig. 4 ).
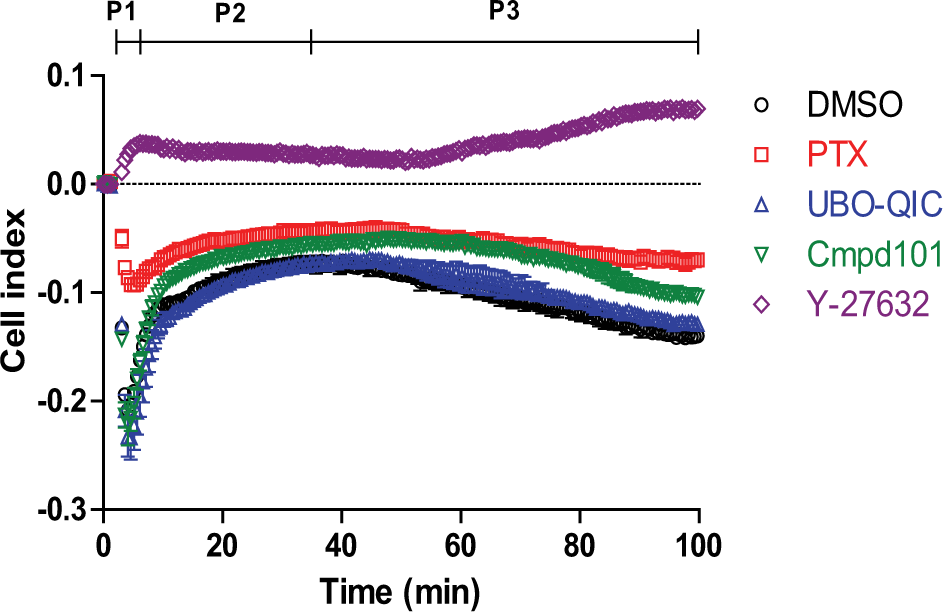
LPA-induced impedance changes in RH7777-LPA1 cells. Changes in impedance in RH7777-LPA1 cells were measured over time following treatment with 100 nM LPA in the absence and presence of 100 ng/mL PTX, 1 µM UBO-QIC, 30 µM Cmpd101, or 30 µM Y-27632. Cells were pretreated with PTX overnight and with other compounds for 30 min prior to LPA stimulation. LPA alone caused rapid decay (P1), followed by rapid ascension (P2) and then slow decay (P3). Data are presented as means ± SEM (n = 3).
Effects of LPA1 NAMs on Impedance
As shown in Figure 4 , LPA-induced changes in impedance are governed by various intracellular signals. Hence, to further characterize LPA1 NAMs, the effects of LPA1 NAMs on impedance were examined and compared with those of Ki16425 and signal inhibitors. Ki16425 almost completely abolished LPA-induced changes in impedance ( Fig. 5A ). Conversely, LQ1–5 had limited effects on the rapid decay phase of LPA-induced impedance. However, the shape of the ascending phase differed between compounds, and although LQ1–3 affected the ascending phase, LQ4 and LQ5 did not. All compounds inhibited the slow decay phase ( Fig. 5B–F and Table 1 ).
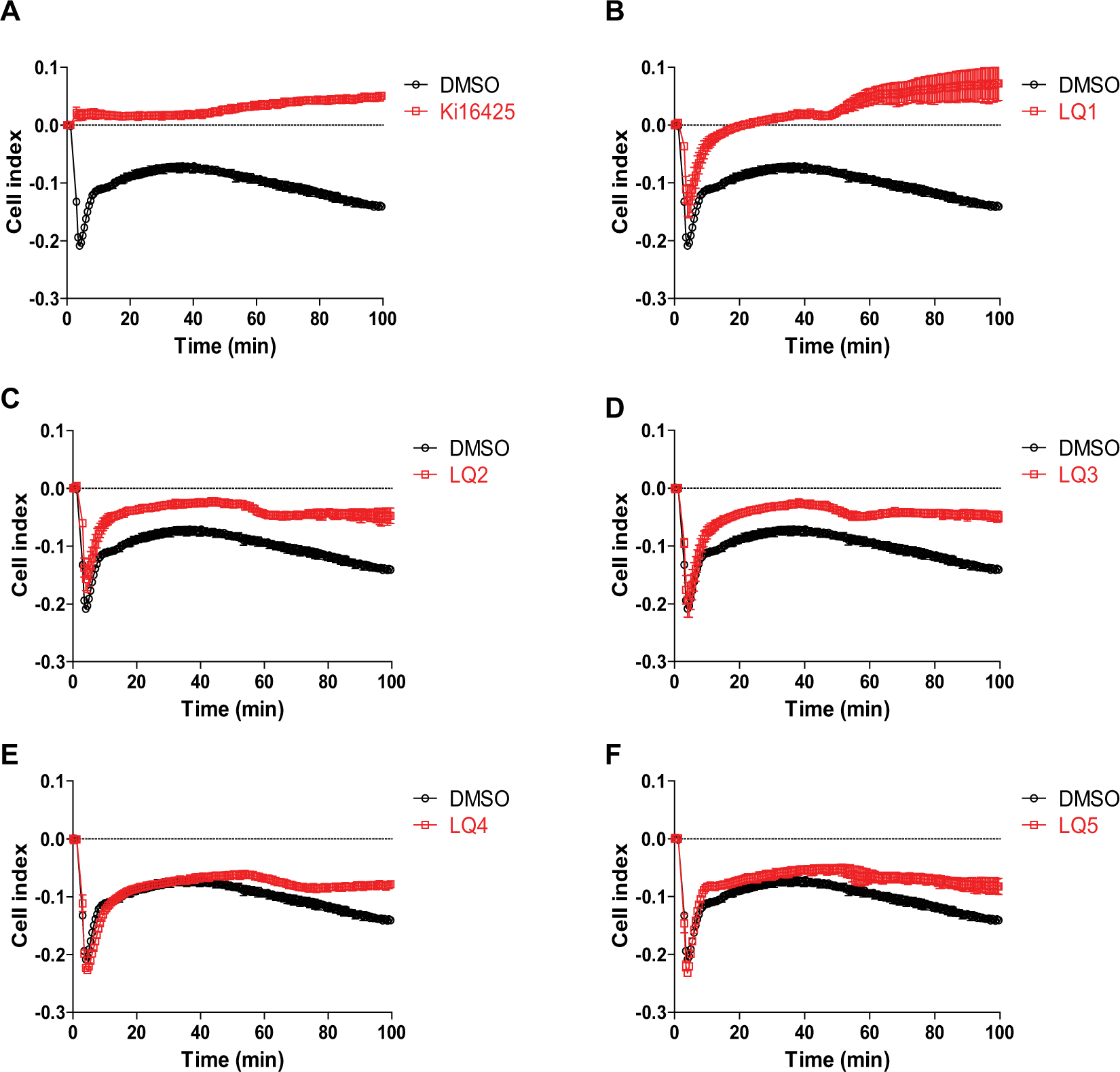
Effects of LPA1 NAMs on LPA-induced changes in cellular impedance. RH7777-LPA1 cells were pretreated with each compound at 30 µM for 30 min and were then stimulated with 100 nM LPA, and impedance changes were measured over time. Data are presented as means ± SEM (n = 3).
Discussion
LPA1 is a multifunctional GPCR that mediates various intracellular signals via Gq, Gi, and G12/13. Although the function of LPA1 has been widely studied, the understanding of the contributions of each signal to cellular function remains poor owing to the complexity of LPA1-mediated signaling pathways. Biased ligands are currently attracting considerable interest because of their potential pertaining to the development of improved drugs and pharmacological tools. Accordingly, we sought to find a tool compound that targets the receptor, and while screening for the compound using multiple assays, we identified five compounds as Gq-biased LPA1 NAMs. These NAMs showed little effect on cAMP responses and β-arrestin recruitment. Among these, LQ4 was the most potent ligand, with an IC50 of 17 nM in Ca2+ flux assays, and was comparable with Ki16425, which had an IC50 of 57 nM.
In this study, we used label-free impedance assays to characterize LPA-induced intracellular signaling. This study was conducted using rat hepatoma RH7777 cells because this cell line is devoid of LPA receptors. 28 Expression of LPA1 enabled us to examine LPA1-specific cellular responses and LPA-induced dynamic impedance changes via LPA1 ( Fig. 4 and Suppl. Fig. S3 ). Moreover, impedance measurements in the presence of inhibitors of G-proteins and kinases that are associated with GPCR activation revealed contributions of each signal to changes in cellular impedance. Previous studies suggested that the G12/13-mediated Rho/ROCK pathway and the Gi-mediated pathway are involved in cell mortality and cytoskeletal changes13,29 and morphological changes and migration, 30 respectively. In agreement, the ROCK inhibitor Y-27632 greatly affected LPA-induced changes in impedance and Gi inhibitor PTX distinctly modified LPA-induced impedance ( Fig. 4 ). Conversely, although Gq signals have been identified as primary contributors to vascular smooth muscle cell migration and proliferation, 9 Gq inhibition slightly affected LPA-induced impedance changes in RH7777-LPA1 cells. These observations suggested that cell migration and/or intracellular signaling markedly differ between cell types and based on conditions of cells and components of extracellular matrices.
LQ1–5 were also characterized using label-free impedance assays, and the effects of Ki16425 and LQ1–5 on LPA-induced changes in impedance differed ( Fig. 5 ). In particular, Gq-biased LQ1–5 had little effect on LPA-induced changes in the rapid decay phase. Taken together with the observation that the ROCK inhibitor Y-27632 inhibited the rapid decay phase, these results suggest that LQ1–5 have no effect on the G12/13/ROCK pathway. Further studies using more direct functional assays to detect G12/13 activities, such as GTPγS binding assays, will draw further conclusions on biased signaling profiles of LQ1–5. The difference was also observed among the Gq-biased NAMs: LQ1–LQ3 increased changes in impedance in the ascending phase, whereas LQ4 and LQ5 did not ( Fig. 5B–F ), suggesting that these compounds differently modulate LPA1. On the other hand, LQ1–5 inhibited the slow decay phase. This property may reflect the involvement of unexpected signaling biases other than those of G12/13 or Gi because the impedance profiles of LQ1–5 were distinct from those of ROCK and Gi inhibitors ( Fig. 4 ).
Because impedance changes may depend on the cell type, observed differences in recombinant RH7777-LPA1 cells may not be directly comparable under physiological conditions. However, these differences may suggest that these compounds stabilize different active conformations of the receptor. In addition to differing profiles of cellular impedance, differing effects on LPA DRCs (Emax lowering effect vs. simple rightward shift effect, Fig. 3 and Table 1 ) also suggested the presence of various active receptor states. Hence, these compounds may produce distinct cellular phenotypes under physiological conditions, warranting further studies on the effects of different types of Gq-biased NAMs in various cells using various assessments of phenotype, such as those of cell migration, proliferation, and survival. Moreover, crystal structures of LPA1 in the presence of these different types of ligands are highly desired to further understand the active states of the receptor.
In summary, we identified novel Gq-biased LPA1 NAMs with multiple chemical scaffolds and characterized these using label-free impedance assays. These compounds provide new pharmacological tools to study the functions of LPA1 in greater detail and may contribute to the development of improved drugs.
Footnotes
Acknowledgements
We thank Shoichi Okubo and Junichi Miyazaki for preparing plasmid and RH7777-LPA1 cells, respectively. We thank Masayuki Kobayashi for conducting HTS. We also thank Yasufumi Miyamoto for synthesizing Cmpd101 hydrochloride. We would like to acknowledge Osamu Sano for helpful discussions and Tomohiro Kawamoto for his support of this study.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.