
Abstract
Huntington’s disease (HD) is a neurodegenerative disease caused by an expansion of CAG trinucleotide repeat (polyglutamine [polyQ]) in the huntingtin (HTT) gene, which leads to the formation of mutant HTT (mHTT) protein aggregates. In the nervous system, an accumulation of mHTT protein results in glutamate-mediated excitotoxicity, proteosome instability, and apoptosis. Although HD pathogenesis has been extensively studied, effective treatment of HD has yet to be developed. Therapeutic discovery research in HD has been reported using yeast, cells derived from transgenic animal models and HD patients, and induced pluripotent stem cells from patients. A transgenic nonhuman primate model of HD (HD monkey) shows neuropathological, behavioral, and molecular changes similar to an HD patient. In addition, neural progenitor cells (NPCs) derived from HD monkeys can be maintained in culture and differentiated to neural cells with distinct HD cellular phenotypes including the formation of mHTT aggregates, intranuclear inclusions, and increased susceptibility to oxidative stress. Here, we evaluated the potential application of HD monkey NPCs and neural cells as an in vitro model for HD drug discovery research.
Introduction
Neurodegenerative diseases affect millions of patients and their families. Because of the complexity of disease pathogenesis, the development of treatments that could slow down the progression of these diseases has been a challenging task. Huntington’s disease (HD) is a fatal neurodegenerative disorder caused by trinucleotide repeat expansion. The expansion of cytosine-adenine-guanine (CAG) repeats in the first exon of the HTT gene on chromosome 4p16.3 results in the development of HD. An individual with more than 36 CAG repeats will develop HD. The age of onset is inversely correlated with the size of the CAG tract, whereas the severity of disease is directly correlated with the CAG repeat length. 1 Drugs and small molecules are expected to elicit systemic benefit on HD phenotypes. Besides the challenge in developing effective therapeutics, the lack of a preclinical large animal model has limited the option for assessing the efficacy of novel therapeutics with outcome measurements similar to those used in humans. In fact, most of the HD rodent models capture only a fragment of HD phenotypes and are difficult to use for long-term assessment because of their short life span. The recent development of an HD monkey model that developed HD clinical features with progression similar to human HD patients suggested the potential of this unique model system for preclinical study.2,3 Because the HD monkey model holds great promise as a preclinical animal model of HD, the current study has evolved based on the recent development of HD monkey neural progenitor cells (HD-NPCs) and their derivative neural cells (HD-NCs) that developed HD cellular features including increased susceptibility to oxidative stress, increased apoptosis, the formation of mutant HTT (mHTT) aggregate, and intranuclear inclusions. 4 To further determine if HD-NPCs and HD-NCs derived from pluripotent stem cells (PSCs) of HD monkeys are a potential in vitro platform for HD drug discovery research, we examined the effect of three known drugs (rilizole [2-amino-6-trifluoro-methoxy-benzothiazole; RI], memantine [1-amino-3, 5-di-methyl-adamantane; ME], and methylene blue [MB]) that had shown beneficial effects on HD. Cytotoxicity, apoptosis, and mHTT aggregation were used as outcome measurements to determine if HD cellular pathologies were improved. Here, we hypothesize that HD-NPCs and HD-NCs can be used as an in vitro platform for drug discovery research of HD.
Materials and Methods
NPC Culture and Neural Differentiation
WT-2 (rZH-2: wild-type [WT] embryonic stem cells [ESCs]), 4 WT-14 (RiPS-14: WT induced pluripotent stem cells [iPSCs]), 4 HD-2 (RPg-2: HD ESCs), 5 and HD-3 (RiPS-3: HD iPSCs) 6 – derived NPCs were used in this study. 4 NPC derivation and culture were described previously. 4 In brief, NPCs were cultured with neural proliferation medium, which was composed of Neurobasal medium (Life Technologies, Carlsbad, CA), 1x B27 (Life Technologies), 2 mM L-glutamine, basic fibroblast growth factor (R&D Systems, Minneapolis, MN, 20 ng/mL), and mouse leukemia inhibitory growth factor (Chemicon, Darmstadt, Germany, 10 ng/mL) on a tissue culture dish coated with polyornithine/laminin (P/L; Sigma, St. Louis, MO). NPCs were passaged by using Stem ProAccutase (Life Technologies).
WT-2, WT-14, HD-2, and HD-3 NPCs were differentiated in vitro by using an established protocol. 4 In brief, WT-NPCs and HD-NPCs were seeded onto a P/L-coated tissue culture dish in 30,000 cell/cm2 concentration. On the next day, neural differentiation medium, composed of DMEM/F12 (Life Technologies), 1x P/S (Invitrogen, Carlsbad, CA), 2 mM L-glutamine, 1x N2 (Life Technologies), 1x B27 (Life Technologies), and 0.1 mM 2-Mercaptoethanol (Sigma) were cultured for 4 days. Then, 0.2 µg/mL sonic hedgehog (R&D Systems) and 0.1 µg/mL fibroblast growth factor–8 (R&D Systems) were then added into differentiation medium. Ascorbic acid (200 mM, Sigma) was added into the differentiation medium on day 8 and cultured until day 21, when differentiated neural cells were collected for analysis.
To determine drug response, NPCs and NCs were treated with 10 µM RI (Sigma), 10 µM ME (Sigma), and 0.1 µM MB (Sigma) for 24 h prior to analysis. 7 Except for MB treatment, 0.1 µM MB (Sigma) was supplemented in the last 7 days of differentiation. 8 All samples were collected for viability, cytotoxicity, and apoptosis analysis.
Immunocytochemistry
NPCs and NCs cultured on P/L-coated glass slides were fixed in 4% paraformaldehyde after treatment with candidate drugs. Fixed cells were permeabilized and blocked with 0.2% Triton-X-100 (Sigma) and 3% bovine serum albumin (Sigma) in phosphate-buffered saline (PBS). Fixed slides were incubated overnight at 4 °C with primary antibodies. Slides were washed twice in PBS followed by incubation with secondary antibody for 1 h at room temperature. Images were taken by using a fluorescence microscope equipped with CellSens software (Olympus, Tokyo, Japan). To quantify the percentage of positive cells, five pictures were captured randomly and counted in two biological replicas by examiner with no knowledge of the samples. A total of 1000 to 2500 cells were counted for each treatment. Primary and secondary antibodies are listed in Supplementary Table S1 . 4
Real-Time Quantitative PCR
Total RNA from cell samples was prepared by using TRIzol (Life Technologies). Genomic DNA was removed by using the Turbo DNA-free Kit (Life Technologies) according to the manufacturer’s instructions. Total RNA (1000 µg) was reverse transcribed using an RNA-to-cDNA kit (Applied Biosystems, Foster City, CA). Quantitative PCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, Hercules, CA) using the CFX96 Real-Time Detection System (Bio-Rad). Quantitative PCR primer sequences are listed in Supplementary Table S2 .
Western Blot Analysis
After treatment for 24 h (RI and ME) or 7 days (MB), cells were harvested and total protein was extracted by using RIPA buffer with protease inhibitor. The total protein concentration was quantified by Bio-Rad DC Protein Assay (Bio-Rad). An equal amount of protein extract was loaded and separated by electrophoresis in 9% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel. Proteins were transferred onto a polyvinylidene fluoride membrane and probed with primary antibodies mEM48 and γ-tubulin (Millipore, Darmstadt, Germany) at 4 °C overnight followed by thorough washes and incubation with secondary antibody. A positive band was visualized by using SuperSignal West FemtoChemiluminescent Substrate (Thermo Scientific, Waltham, MA) and imaged by ChemiDoc Imaging System (Bio-Rad). 4
Cell Viability and G6PD Cytotoxicity Assay
For cell viability, WT-NPCs or HD-NPCs were plated in a P/L-coated 96-well plate. The next day, NPCs were treated with drugs in culture medium for 24 h. A cell viability assay was performed by MTT assay (ATCC) following the manufacturer’s instructions.
For cell cytotoxicity, WT-NPCs or HD-NPCs (or WT-NCs or HD-NCs) were treated with drugs in culture medium for 24 h. A Vybrant G6PD Cytotoxicity Assay Kit (Life Technologies) was used to determine cytotoxicity according to the manufacturer’s instructions.
For NCs, WT-NPCs or HD-NPCs were differentiated 21 days in 24-well plates coated with P/L. On day 21, fresh media with drugs were replaced followed by 24 h of incubation. Media were then collected and spun down to collect all suspended cells. The cell pellets were then resuspended in G6PD reagent with lysis buffer and added into the culture well. The percentage of G6PD released in media was normalized with the total neural cell lysate from the corresponding well. 4
Statistical Analysis
All experiments were composed of three biological replicas. Statistical analysis was performed by using SPSS 12.0 (SPSS Inc., Chicago, IL). Data were represented as mean ± standard error. Statistical differences were calculated using analysis of variance (ANOVA). Differences were considered significant at *p < 0.05 and **p < 0.01.
Results
Characterization of NPCs and NCs
WT-NPCs and HD-NPCs derived from WT and HD monkey PSCs were used to determine if they could be used for drug discovery research. HD cellular phenotypes including cytotoxicity, apoptosis, and the formation of mHTT aggregates were used as outcome measures to determine if selected drugs can ameliorate and reverse cellular changes in HD-NPCs and HD-NCs. WT-NPCs and HD-NPCs expressed progenitor cell markers, which include Sox-2, Pax-6, Musashi-1, and Nestin ( Suppl. Fig. S1A). The expression of mHTT was significantly up-regulated in the two HD-NPC lines and their differentiated NCs (HD-2 and HD-3) when compared with the two WT-NPC lines and their differentiated NCs after being normalized to endogenous HTT levels (WT-2 and WT-14; Fig. 1A ). Moreover, HD-3 has a much higher expression level of mHTT than HD-2, which was consistent with the prior report ( Fig. 1A ). 4 Western blot analysis of WT-14 and HD-3 NPC and NCs revealed the accumulation of mHTT aggregate in HD-NCs but not in WT-NCs and NPCs ( Fig. 1B ). A G6PD cytotoxicity assay was used to determine cytotoxicity in NPCs and NCs. No difference was found in cytotoxicity between WT and HD NPCs ( Fig. 1C ). However, a significant increase in cytotoxicity was observed in HD-NCs when compared with WT-NCs ( Fig. 1D ). These results suggested the impact of mHTT in cytotoxicity and mHTT protein aggregation in HD cells. Interestingly, both cytotoxicity and mHTT aggregation were significantly increased in HD-NCs when compared with corresponding HD-NPCs. This finding suggested a differential susceptibility to mHTT in NPCs and NCs; thus, variations in response to treatments are also expected in HD-NPCs and HD-NCs.
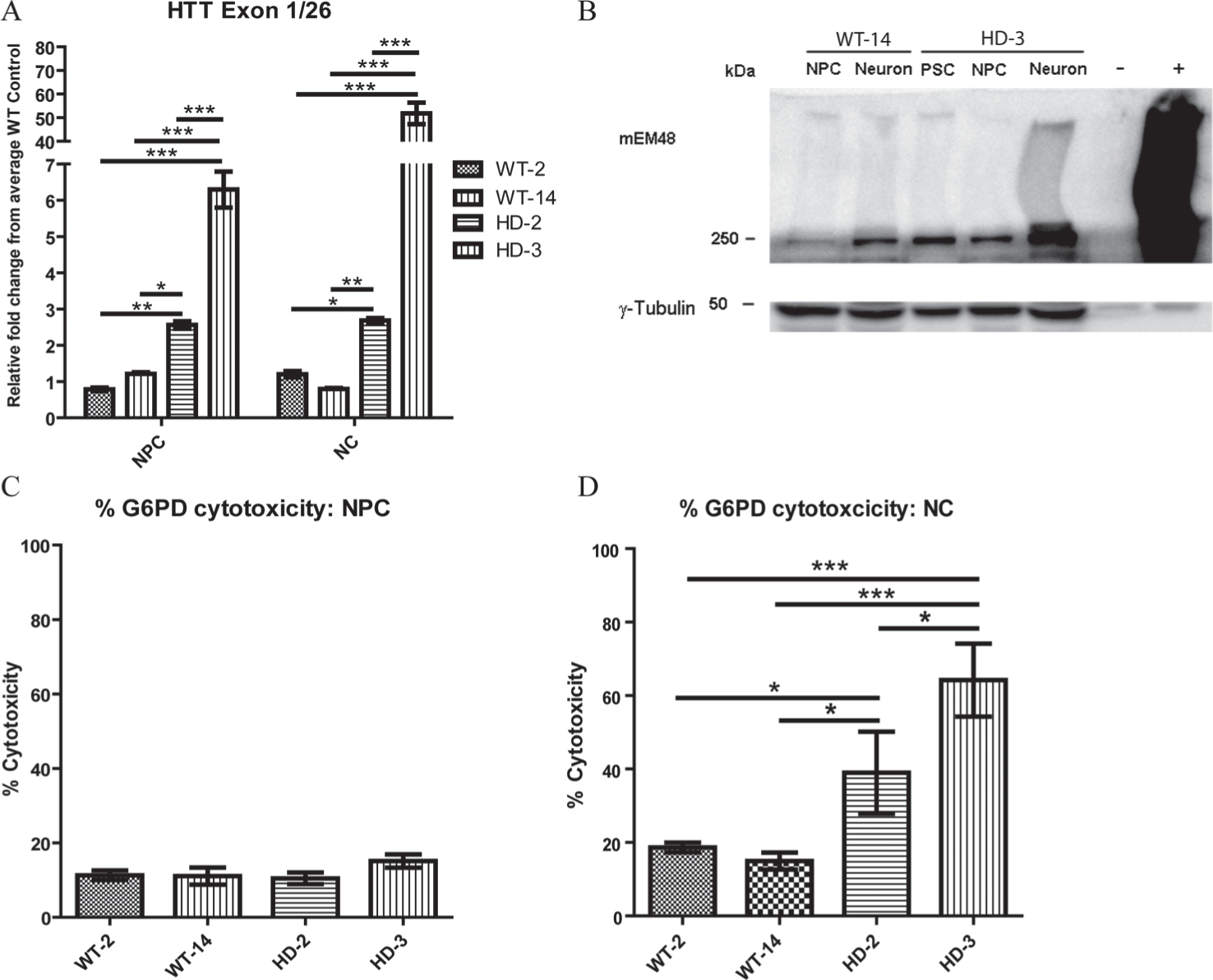
Characterization of wild-type (WT) and Huntington’s disease (HD) neural progenitor cells (NPCs) and differentiated neural cell. (
Assessing Drug Responses in WT-NPCs and HD-NPCs Using an MTT Assay and G6PD Cytotoxicity Assay
RI, 9 ME, 10 and MB 8 were used to treat HD-NPCs and determine if viability and cytotoxicity can be improved. After 24 h of treatment, no significant improvement in viability or cytotoxicity was observed in WT-NPCs or HD-NPCs ( Fig. 2A–H ). This result is consistent with cytotoxicity observed in NPCs in that no significant difference in cytotoxicity was observed when comparing WT-NPCs and HD-NPCs.
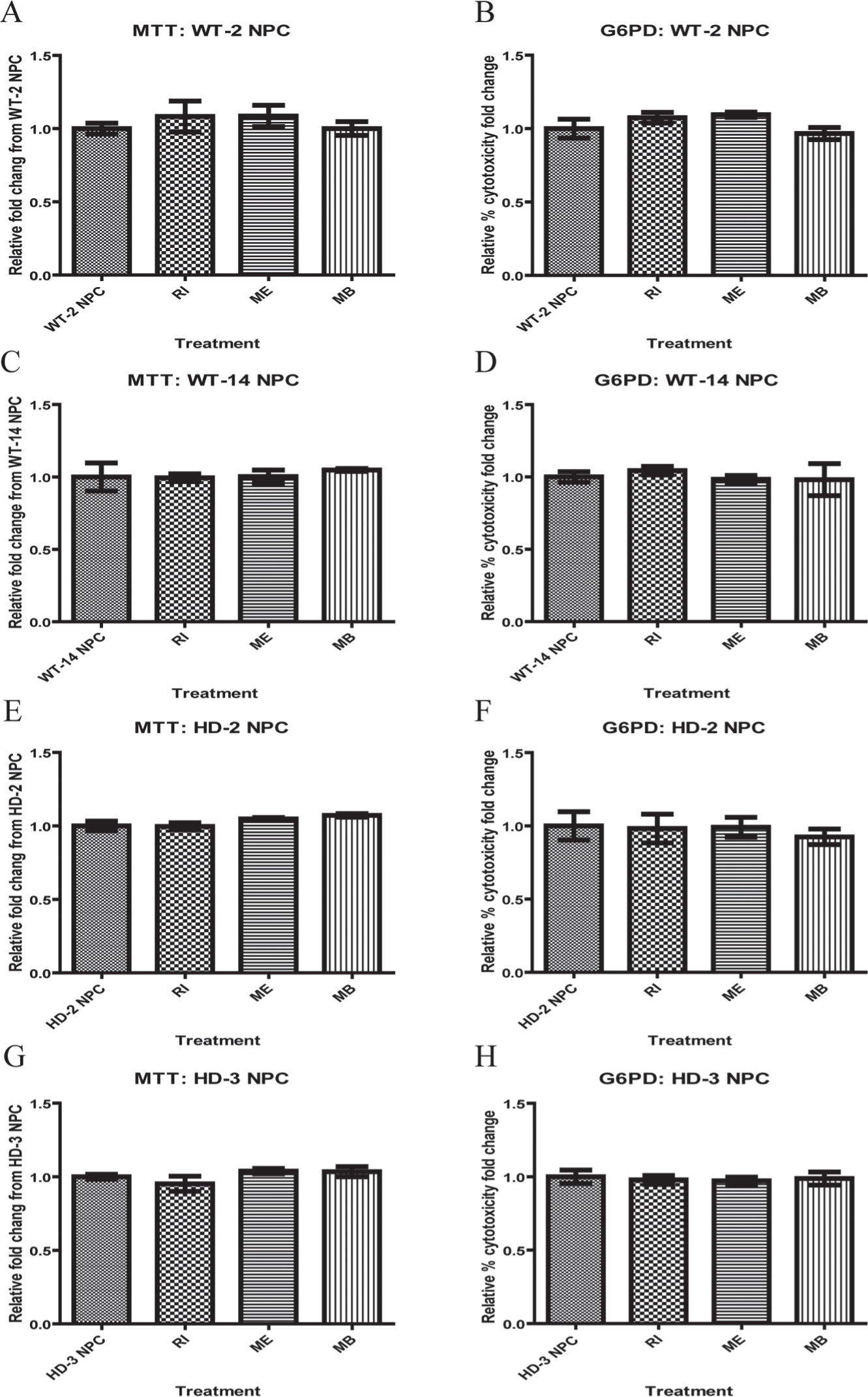
Effect of drugs on wild-type (WT) and Huntington’s disease (HD) neural progenitor cells (NPCs) was determined by using MTT assay and G6PD cytotoxicity assay. (
Assessing Drug Response in WT and HD Differentiated NCs by G6PD Cytotoxicity Assay
WT-NPCs and HD-NPCs were differentiated into NCs followed by treatment with RI, ME, and MB. All differentiated NCs expressed neural-specific markers including Tuj-1, doublecortin (DCX), microtubule-associated protein-2 (Map-2), and Tyrosine hydroxylase (TH) (
Suppl. Fig. S1B
). The formation of intranuclear inclusions and mHTT aggregates was observed in HD-NCs, with a significant increase in cleaved caspase-3 positive cells (
Suppl. Fig. S1B
). RI and ME were administered into culture media on the last day of neural differentiation for 24 h, whereas MB was added into culture media during the last 7 days of differentiation. RI, ME, and MB did not have an effect on NCs derived from the two WT-NPC lines (
Fig. 3A
,
B
). However, significant reduction in cytotoxicity in HD-2 and HD-3 NCs was observed (
Fig. 3C
,
D
, respectively
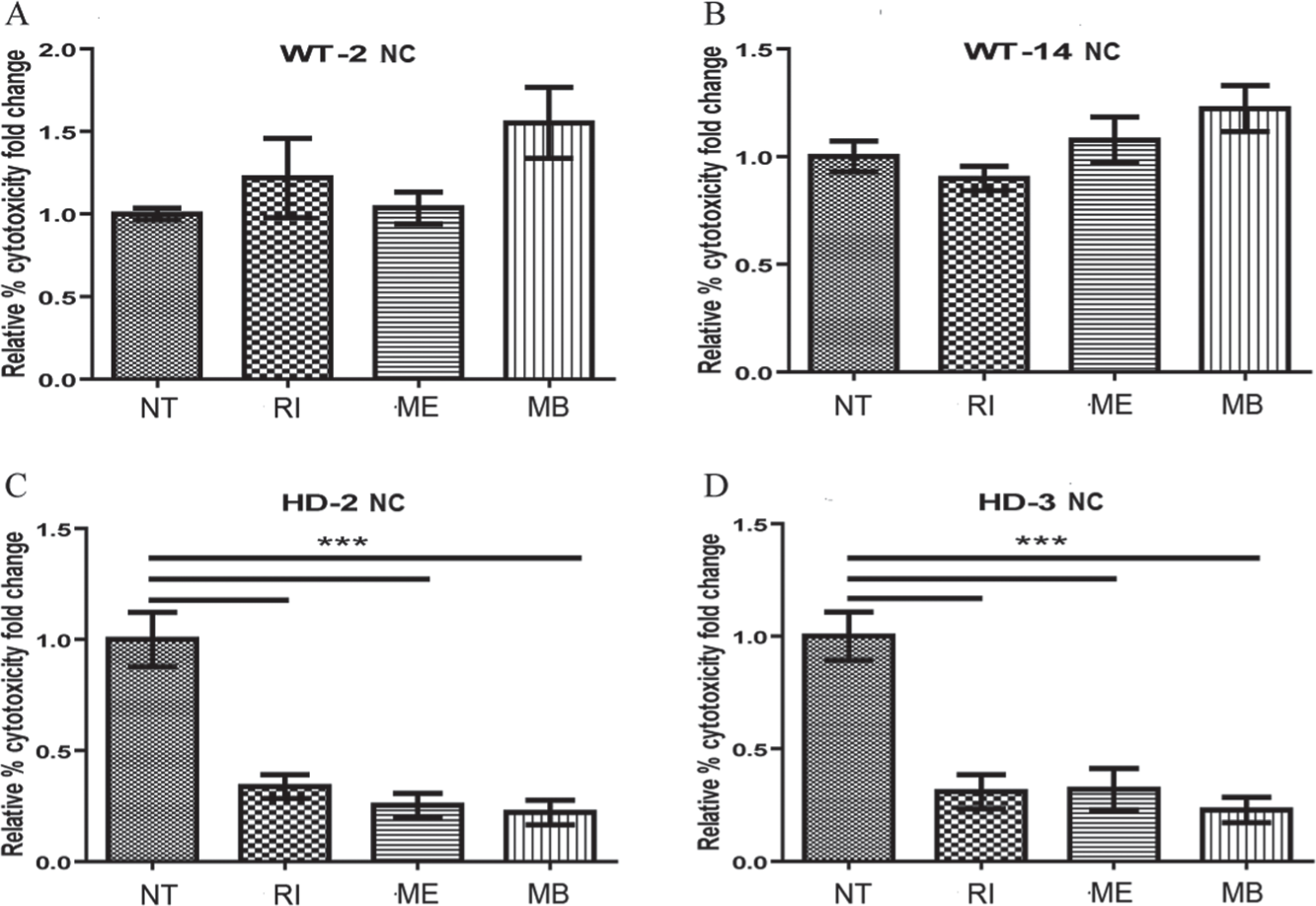
Effect of drugs on wild-type (WT) and Huntington’s disease (HD) differentiated neural cells (NCs) was determined by using a G6PD cytotoxicity assay. Cytotoxicity of WT-2 NC (
Ameliorating Apoptosis and mHTT Aggregation in HD-NCs
Activated caspase-induced apoptosis has been reported as one of the pathological hallmarks in HD. 9 A similar observation was also described in NCs derived from HD-NPCs. 4 To examine if RI, ME, and MB ameliorate apoptotic responses, they were administered into culture media of differentiated WT-NCs and HD-NCs for 24 h followed by fixation and immunostaining using a specific antibody that recognized cleaved caspase-3 and Map-2. The total cell count of Map-2 (green) and cleaved caspase-3 (red) positive cells was used to determine the ratio of the apoptotic NC population ( Fig. 4A–C ). In WT control NCs, the addition of RI, ME, and MB did not affect the number of caspase-3–positive cells or apoptotic NCs ( Fig. 4D ). However, significant reduction in caspase-3–positive cells was observed in HD-NCs (HD-2 and HD-3) treated with RI, ME, and MB ( Fig. 4B–F ). In addition, WT-14 and HD-3 NCs treated with RI, ME, and MB were double immunostained using DCX and mEM48 antibodies to determine if mHTT aggregation was affected in NCs. As expected, no aggregate was observed in WT-NCs with or without treatment ( Fig. 5A ). In contrast, mEM48-positive NCs were reduced in HD-NCs in all treatment groups ( Fig. 5B ), which was further confirmed by Western blot analysis using mEM48 antibody ( Fig. 5C–D ). Between the three drugs, MB has a more robust impact on the reduction of mHTT aggregates as well as a soluble form mHTT protein when compared with RI and ME ( Fig. 5C , D ). These results suggested that RI, ME, and MB can reduce caspase-3–induced apoptosis in HD-NCs, with RI being the most prominent and MB having the least effect. In the case of reducing mHTT aggregation, MB was the most effective whereas ME had no significant impact on both aggregated and soluble forms of mHTT protein. These data were consistent between cytotoxicity, reduced apoptosis, and the formation of mHTT aggregates in HD-NCs.
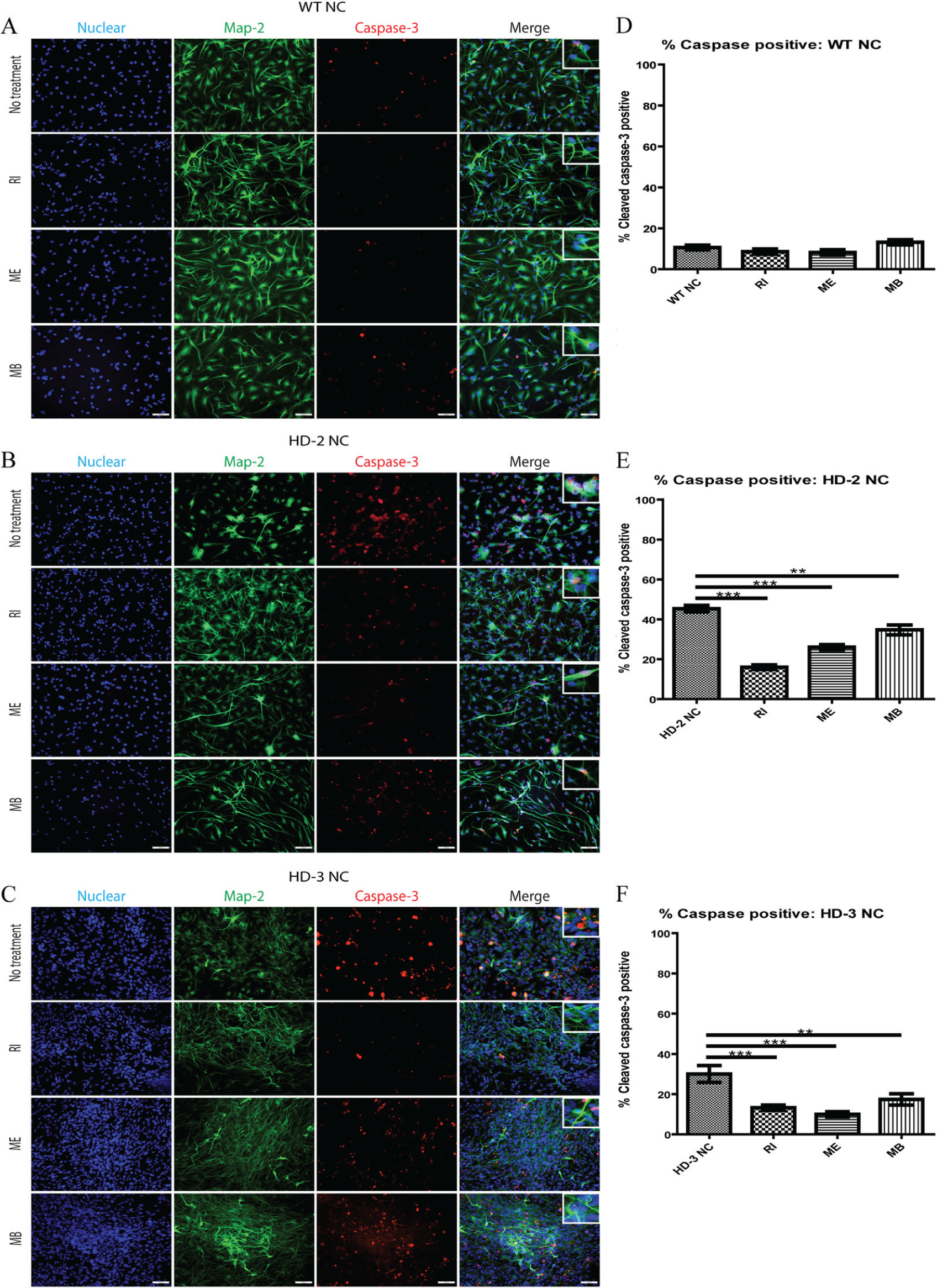
Effect on apoptosis in differentiated Huntington’s disease neural cells (HD-NCs). (
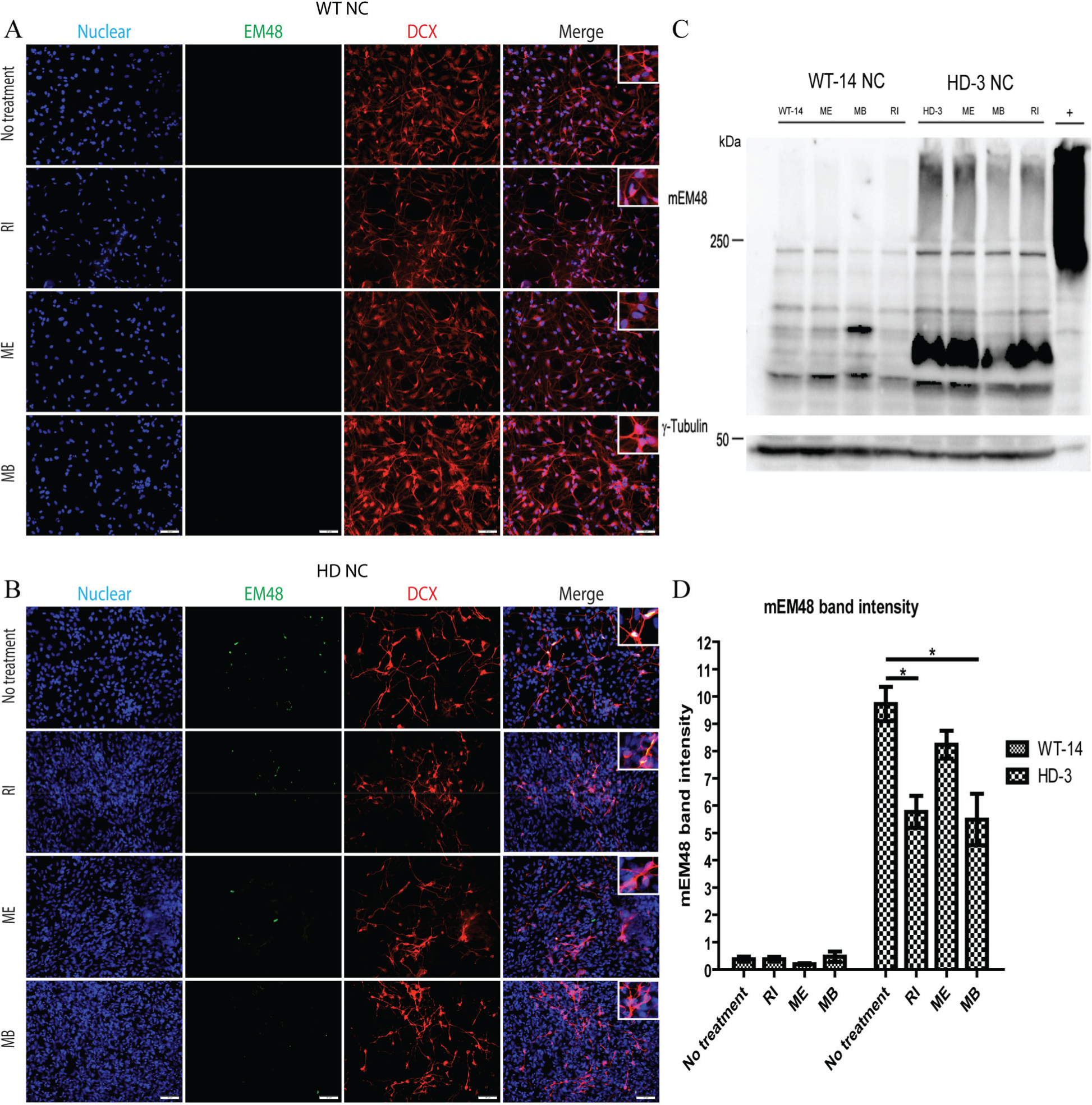
Effect of drugs on mutant HTT (mHTT) protein aggregation in Huntington’s disease neural cells (HD-NCs). (
Discussion
In this study, three drugs that have shown beneficial effects in several neurodegenerative diseases including HD, Parkinson’s disease (PD), and Alzheimer’s disease (AD) were used to determine if HD monkey iPSC-derived NPCs and NCs can be used as an in vitro platform for drug discovery research because of their unique HD cellular phenotypes such as cytotoxicity, apoptosis, and protein aggregation. We have shown that HD-NPCs and HD-NCs are highly susceptible to oxidative stress with HD cellular phenotypes including mHTT aggregates that can be reversed by genetic and biochemical approaches. 4 HD-NCs express glutamate receptors, which are the target of RI and ME, and a positive impact was observed similar to prior studies. 4 HD-NPCs and HD-NCs are unique platforms for drug discovery research in vitro, and HD monkeys can be used as a preclinical animal model for assessing therapeutic efficacy. 3
NPCs and NCs of HD were extremely sensitive to oxidative stress induced by hydrogen peroxide or by the withdrawal of growth factors in culture that resulted in a significant increase in apoptosis.4,11 Compared with HD-NCs, HD-NPCs have a lower mHTT expression level and mild HD cellular phenotypes that may limit its application in drug discovery research because of the lack of robust outcome measurements used to determine the beneficial effect of treatments. Many in vitro platforms have been developed for drug discovery research, including an HD yeast model, 12 brain slice model, 13 genetically modified 293/HEK cell, 14 primary neuronal culture from transgenic rodents, 15 transgenic rodent cell line such as PC12, 16 transgenic NHP cell line, 4 and NCs derived from human iPSCs.17,18 However, similar to HD-NPCs, robust HD cellular phenotypes are very limited in these model systems with a narrow margin for determining therapeutic effect. To the contrary, HD-NCs developed robust cellular phenotypes, including the increase of mHTT aggregates, the formation of intranuclear inclusions, increase in caspase-3–induced apoptosis, and cytotoxicity caused by oxidative stress and the withdrawal of growth factors. Most importantly, HD-NCs can be generated by in vitro differentiation of HD-NPCs. 4
Improvement in cytotoxicity, caspase-3–induced apoptosis, and the reduction of mHTT protein aggregates was observed in HD-NCs treated with RI, ME, and MB. RI has been reported to inhibit ion channels such as glutamate-gated channels, voltage-gated channels, and volume-sensitive chloride channels. 19 RI ameliorates glutamate-mediated excitotoxicity by blocking the voltage-gated sodium channel and inhibits the release of glutamate at the presynaptic terminus. 20 In HD patients, RI enhances neurite formation and growth in damaged motor neurons, reduces the loss of gray matter, and increases serum brain-derived neurotrophic factor. 9 In the YAC128 MSN model, RI significantly reduced glutamate-induced apoptosis. 7 Besides HD, the neuroprotective effect of RI has also been reported in the PD model by attenuating dopaminergic neuron degeneration and suppressed reactive astrocytosis in the striatum. 21 Furthermore, in amyloidtrophic lateral sclerosis (ALS), RI improves the survival rate of ALS patients.22,23 ME is a specific, moderate-affinity, uncompetitive, open-channel N-methyl-D-aspartate glutamate receptor (NMDAR) antagonist. 7 The increased function of NMDAR was observed in the YAC128 mouse model that was caused by selective striatal excitotoxicity and can be prevented by the inhibition of NMDA and mGluR1/5 receptor activity. 24 The elevation of NMDAR activity alters intracellular Ca2+ signaling pathways that lead to cell death, and a synaptic Ex-NMDAR blocker can attenuate mHTT-induced striatal atrophy and motor learning deficits in the YAC128 model. 25 Thus, a glutamate receptor antagonist such as RI and ME has been considered for reducing cytotoxicity in HD, and two open-label clinical trials on these two drugs have shown a neuroprotective effect not only in HD 9 but also in PD 26 and AD. 27
MB belongs to the phenothaizinium family, which has been used in phase IIb clinical trials for the treatment of mild to moderate AD. Significant improvement in cognitive function and slowing down of AD progression has been reported. 28 MB also reduced soluble Aβ and rescued early cognitive deficit in AD transgenic mice. 29 In HD, MB can slow down the aggregation of mHTT protein even when aggregation has already been initiated, and aggregated species were presented in vitro. 8 In primary neurons expressing mHTT, MB reduced the formation of both oligomeric and insoluble mHTT and increased the survival of neurons. 8
Our studies on RI, ME, and MB in HD-NPCS and NCs demonstrated similar beneficial effects in ameliorating HD cellular phenotypes, which were consistent with prior studies in different model systems. 18 Reduction in cytotoxicity and caspase-3–induced apoptosis via glutamate receptor inhibitors such as RI and ME suggested our HD-NPCs and NCs respond to drugs in a similar manner as the other model systems. A similar effect on the reduction of mHTT protein aggregate and the soluble form mHTT protein was also observed in HD-NCs treated with MB and RI.8,20 In a 3-nitropropionic–induced HD model, ME was shown to reduce HTT levels, which further suggested its potential in reducing the proteolytic HTT fragment. 30 However, a similar suppression effect in mHTT by ME has not been reported in other model systems, which is consistent with our findings ( Fig. 5C ). To demonstrate the effect on mHTT aggregation, we chose to use HD-3 because more robust mHTT aggregation has been observed. 4 HD-3 has 72 CAG repeats, whereas HD-2 has only 29 CAG repeats with a much lower mHTT aggregation that limited it from assessing the impact on mHTT aggregation. Although the mechanism and therapeutic effect of RI, ME, and MB in HD have yet to be determined, our HD-NPCs and NCs offered not only a unique alternative platform for understanding the underlying mechanism of these effective drugs but also a novel platform with robust phenotypic outcome measures for drug discovery research in HD.
In conclusion, this study is not aimed at discovering novel drugs for HD but rather validates as a proof of principle the potential application of HD-NPCs and NCs in drug discovery research by using drugs that have been known for their therapeutic benefits in neurodegenerative diseases such as HD, PD, and AD. Although our prior study has suggested the potential of HD-NPCs and NCs in drug discovery research based on the positive effect of ME in ameliorating cytotoxicity, in-depth validation using different assessment tools and methods was not performed. 4 Here, we perform direct comparison of the protective effect of three known drugs using multiple assays to confirm if HD-NPCs and NCs can effectively respond to drugs that have shown benefits for neurodegenerative diseases. Although HD-NPCs and HD-NCs can serve as an in vitro drug discovery platform, HD monkeys with similar genetic constitution as the HD-NPCs and HD-NCs can be used for preclinical assessment prior to a preclinical study using HD monkeys followed by a clinical trial in humans to further ensure the safety and potential benefit of the new treatment.2,3
Footnotes
Acknowledgements
All experimental procedures described in this study were approved by Emory Environmental Health and Biosafety committee. We thank Ms. Leslee Sinclair for her assistance in proofing and editing. Tanut Kunkanjanawan and Rangsan Pranpai are supported by the Royal Golden Jubilee PhD program of Thailand Research Fund. YNPRC is supported by the Office of Research and Infrastructure Program (ORIP)/OD P51OD11132. This study is supported in part by grant awarded by the NINDS (NS084163) and the ORIP/NIH (OD010930; Transgenic Huntington’s Disease Monkey Resource) to AWSC.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.