
Abstract
Thrombotic microangiopathy (TMA) is a rare but potentially life-threatening complication following autologous stem cell transplantation (ASCT). The incidence is extremely low. Here, we present the case of a 54-year-old male with relapsed diffuse large B-cell lymphoma (DLBCL) who developed TMA within 92 days after ASCT. The patient exhibited signs of hemolysis, thrombocytopenia and rapid renal function deterioration, and necessitating initiation of hemodialysis. Kidney biopsy confirmed TMA and Genetic testing revealed Complement H factor deficiency. Treatment with plasma exchange and corticosteroids resulted in no significant response. The patient was ultimately managed with ravulizumab. Transplant associated TMA (TA-TMA) is a an entity in which TMA occurs following stem cell transplant. This case highlights the importance of considering TMA in the differential diagnosis of renal failure following ASCT and initiating prompt treatment as delayed management can lead to devastating outcomes.
Introduction
Thrombotic microangiopathic hemolytic anemia (TMA) is a rare but potentially life-threatening complication characterized by microvascular thrombosis, hemolytic anemia, and thrombocytopenia. 1 While TMA is most commonly associated with conditions such as thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS), it can also occur in the setting of stem cell transplantation. 2 When TMA occurs post-stem cell transplantation, it usually happens after allogeneic rather than autologous stem cell transplant. 3 The pathophysiology of TMA in autologous stem cell transplant is multifactorial and involves complex interactions between transplant-related factors and patient-specific variables. Endothelial injury, immune dysregulation, and genetic predisposition may contribute to the development of TMA in this context. 4
Complement Factor H (FH) is a crucial protein in the complement system, responsible for regulating the alternative pathway. Complement Factor H deficiency can lead to conditions like atypical hemolytic uremic syndrome and C3 glomerulopathy. 5 Genetic studies can help identify rare causes of TMA, and testing may be necessary when the diagnosis is unclear. 6 Diagnosing TMA can be challenging and is sometimes made retrospectively or on kidney biopsy.
Case presentation
A 54-year-old male with a 24-year history of antiphospholipid syndrome and DVT, treated with anticoagulation, and relapsing diffuse large B-cell lymphoma, who had undergone BEAM conditioning for an autologous stem cell transplant 3 months prior, presented to the hospital with abdominal pain, nausea, and vomiting. A CT scan of the abdomen with intravenous contrast revealed acute cholecystitis. He was managed conservatively with IV antibiotics and a percutaneous cholecystostomy and was discharged home for follow-up.
He presented again from the outpatient clinic with evidence of acute kidney injury (AKI). He was asymptomatic at the time, and his physical examination showed a blood pressure of 152/95, with the rest of the examination being unremarkable. Platelet count was slightly lower than his baseline. Initially, as he presented 2 days after the IV contrast, it was thought to be contrast associated nephropathy. He was managed accordingly and remained non-oliguric and well-compensated throughout. Urinalysis revealed proteinuria and RBCs.
His kidney function failed to improve, an expanded AKI workup was initiated, which showed negative ANA and ANCA and normal complement levels (low C3).Table 1. Antiphospholipid syndrome (APS) titers were high, raising concerns for catastrophic APS apheresis was carried out which resulted in no improvement. Due to the lack of improvement in his kidney function, a kidney biopsy was performed.
Laboratory evaluation.

Kidney biopsy showed non-proliferative mesangial stalk widening with narrowing of capillary lumina. Silver stain revealed mesangiolysis and segmental glomerular basement membrane duplication. Preglomerular arterioles demonstrated fibrinoid necrosis. There was focal interstitial hemorrhage, fibrin exudates, and tubular necrosis. Immunofluorescence was negative for immunoglobulin deposition. Electron microscopy revealed endothelial cell swelling, widening of the lamina rara interna, and focal basement membrane duplication with entrapped cytoplasmic fragments; there were no immune complex type electron dense deposits. The features were consistent with thrombotic microangiopathy (TMA; Figures 1 and 2) The apheresis team was contacted, and he underwent one session of apheresis along with IV methylprednisolone pulse steroid, but there was no improvement.
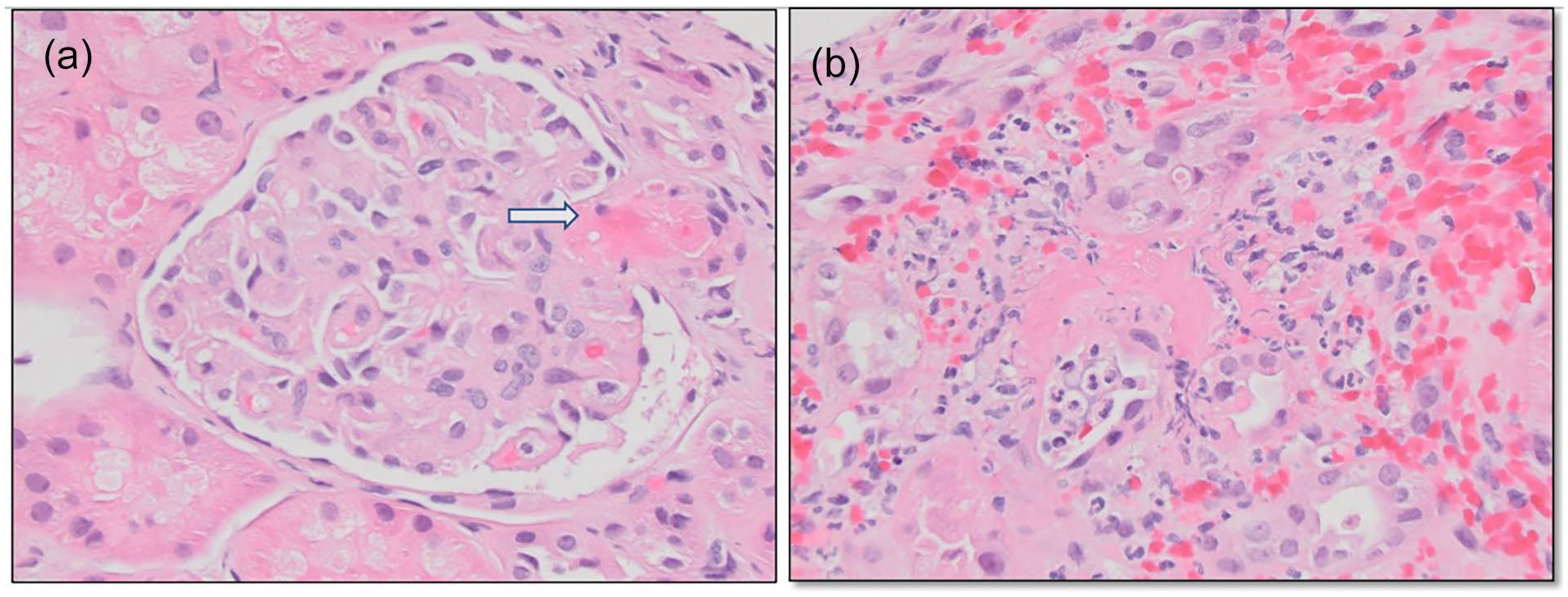
(a) Fibrinoid necrosis of pre-glomerular arteriole (arrow). Tubular epithelium shows cytoplasmic vacuolization (H&E, ×400). (b) Tubular injury and necrosis which interstitial hemorrhage, fibrin exudates, and polymorphonuclear leukocytes (H&E, ×400).
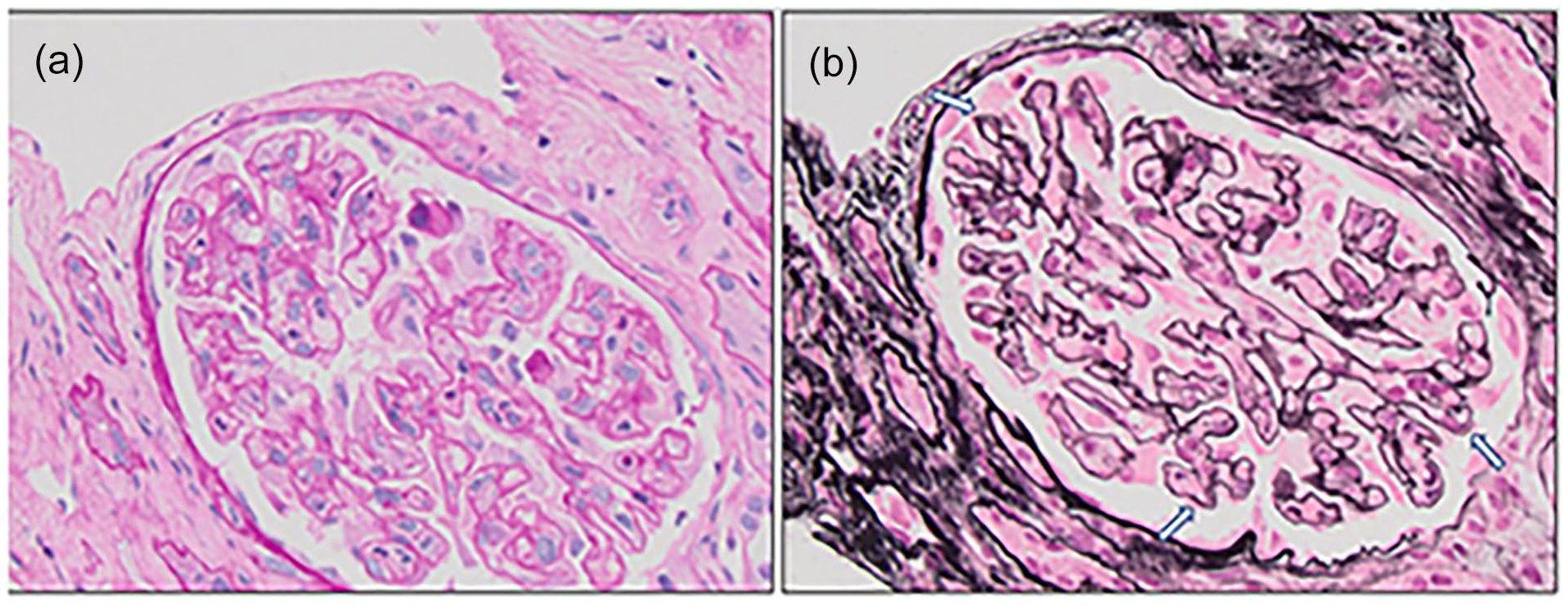
(a) Mesangiolysis and capillary wall thickening (PAS, ×400). (b) New basement menbrance formation (“reduplication”) duplication of the basement membrane (arrows) and mesangiolysis (Jones methenamine silver, ×400).
Genetic testing was conducted and revealed a complement factor H (CFH) deficiency. He remained dialysis-dependent and was discharged on dialysis. Seen by hematology as an outpatient, genetic studies ultimately revealed a homozygous deletion of CFHR4-CFHR1, which is associated with antibodies to factor H and an increased risk of developing atypical hemolytic uremic syndrome (HUS), consistent with CFH deficiency. It was decided to start him on ravulizumab with two loading doses, followed by maintenance doses every 8 weeks for a total of 6 months with an ultimate plan to continue raviluzumab for a total of one year if given continued improvement with treatment.
He remains dialysis-dependent as his kidney function showed no signs of recovery; however, his blood parameters improved. As well as improved BP measures.
Discussion
The development of thrombotic microangiopathy (TMA) in the context of autologous stem cell transplantation (ASCT) is a multifactorial process. 7 Conditioning regimens used in ASCT, comprising high-dose chemotherapy and sometimes total body irradiation, can induce extensive endothelial damage, disrupting the balance between procoagulant and anticoagulant factors on the endothelial surface. This endothelial injury predisposes patients to microvascular thrombosis and subsequent hemolysis, hallmark features of TMA. 8
Furthermore, the infusion of autologous stem cells post-transplantation may elicit immune reactions, exacerbating endothelial injury, and inflammation. 9 Immune-mediated endothelial damage can be triggered by donor-derived T cells recognizing recipient tissues as foreign, leading to a graft-versus-host-like reaction. 10 Additionally, proinflammatory cytokines and chemokines released from infused stem cells contribute to the inflammatory milieu, further promoting endothelial dysfunction and TMA pathogenesis. 11
The incidence of TA-TMA is largely unknown, ranging from 0.5% to 76% following allogenic stem cell transplants. In contrast, the incidence is much lower after autologous transplants, up to 27%. 12 Most cases of TA-TMA after autologous transplants occur primarily in pediatric patients, with the incidence in adults remaining unclear. 13 The median time for developing this condition in the pediatric population is approximately 35 days post-transplant. 14 However, in adults it was mainly seen after 3 months following stem cell transplant.
The microscopic findings in TMA are similar regardless of the disease’s etiology. Some features, however, may be more characteristic of a specific etiology. Severe mesangiolysis as found in this case, is characteristic of TMA associated with hematopoietic stem cell transplantation. 15 Typical microscopic features in TMA, are those of severe endothelial injury, including glomerular endothelium,and may show any of the following features: thrombosis, fibrinoid necrosis, capillary wall thickening due to endothelial cell swelling and mesangial interposition with new basement membrane formation, luminal narrowing or occlusion, mesangiolysis, and acute inflammation. 16 Pre-glomerular arterioles may demonstrate endothelial swelling, fibrinoid necrosis, and fragmented red blood cells or thrombi. Recanalization of thrombus produces “glomeruloid” lesions. On immunofluorescence, fibrinogen positivity is expected in the walls of arterioles and small arteries, and may be present in the glomeruli. Glomeruli may exhibit non-specific staining for immunoglobulins. On electron microscopy, one of the most characteristic ultrastructural findings of TMA is thickening of the glomerular capillary walls due to endothelial cell swelling and lifting with mesangial interposition. 17
One notable genetic factor implicated in the pathogenesis of TMA is complement factor H (CFH) deficiency. 18 CFH is a key regulator of the alternative pathway of the complement system, which plays a crucial role in innate immunity and inflammation. Genetic mutations affecting CFH, along with other complement regulatory proteins such as factor I (CFI) and membrane cofactor protein (MCP), have been associated with an increased risk of TMA development. 19
Our patient was found to have CFH deficiency mutation. CFH deficiency remains an important consideration in patients with TMA following ASCT, especially in those with a family history of complement-mediated disorders or recurrent TMA episodes. Alongside other complement regulatory proteins like factor I (CFI) and membrane cofactor protein (MCP), CFH deficiency disrupts the delicate balance of the complement cascade, leading to uncontrolled complement activation, endothelial cell damage, and subsequent microvascular thrombosis.
The presence of CFH deficiency mutation points toward the need for targeted therapy to inhibit complement activation and mitigate TMA-associated complications. Monoclonal antibodies such as eculizumab and ravulizumab, which specifically target complement protein C5, have demonstrated efficacy in treating TMA associated with CFH deficiency. 20 By blocking the terminal complement pathway, these antibodies prevent the formation of the membrane attack complex (MAC), attenuating hemolysis, and endothelial damage. 21
Catastrophic antiphospholipid syndrome (CAPS) was considered for our patient, but it was ruled out. The diagnosis of CAPS requires meeting specific criteria: positive antibodies, evidence of small vessel occlusion, and involvement of two or more organ systems occurring simultaneously within a week. Small vessel occlusion typically shows evidence of both thrombosis and vasculitis. 22 Although our patient had evidence of thrombosis on kidney biopsy, there was no indication of vasculitis. He underwent one session of plasma exchange (PLEX) as a possible treatment for CAPS, which did not alleviate his symptoms. Ultimately, he improved with terminal complement blockade.
The decision to initiate treatment with ravulizumab in our patient reflects the recognition of CFH deficiency as a critical determinant of TMA pathogenesis post-ASCT. We believe that TMA was triggered by acute cholecystitis as part of the second hit phenomenon.
However, while targeted complement inhibition with ravulizumab represents a promising treatment, several considerations are necessary. These include the optimal timing of treatment initiation, dosing regimen, and duration of therapy. 23 Additionally, close monitoring for treatment response and adverse effects is essential to ensure optimal patient outcomes. Ravulizumab, a long-acting C5 inhibitor, offers the advantage of extended dosing intervals compared to eculizumab, potentially enhancing patient convenience and treatment adherence. 24
Conclusion
Thrombotic microangiopathic hemolytic anemia is a rare but potentially life-threatening complication following autologous stem cell transplantation. Allogeneic stem cell transplant related TMA remains more common than Autologous SCT related TMA. Prompt recognition and management are essential for improving outcomes in affected patients. Genetic factors have been noted to trigger TMA in susceptible patients. Treatment options include plasma exchange, corticosteroids, and complement inhibitors. Further research is needed to better understand the pathophysiology of TMA in the context of ASCT.
Footnotes
Acknowledgements
We thank the nephrology department at penn state health for giving us the opportunity and support to conduct this work
Author contributions
Ahmad Matarneh: Manuscript writing, literature review, Clinical care; Sundus sardar Manuscript writing, literature review, Clinical care; Lidys rivera galvis: Pathology, manuscript writing; Catherine Abendroth: Pathology, manuscript writing; Naman Trivedi: Manuscript writing, literature review, Clinical care; Mathew Evans: Manuscript writing, literature review, Clinical care; Nasrollah ghahramani: Clinical care, literature review, manuscript write up, Mentor.
Date availability statement
The data that support the findings of this study are available on request from the corresponding author.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The funding was provided from Pennsylvania state university.
Ethical approval
N/A. not required.
Gurantor
N.G.
Trial registration
N/A.