
Abstract
Kidney disease is a frequent complication after haematopoietic stem cell transplant (SCT), with glomerulopathies reported in up to 1–6% of allogeneic SCT recipients. Membranous nephropathy is the commonest reported, followed by minimal change nephropathy. Recent work has identified a target antigen in post-SCT membranous nephropathy. We describe seven cases of post-SCT nephrotic syndrome due to de novo glomerulopathy. In the changing landscape of post-SCT glomerulopathy, we review the literature and describe how our cases demonstrate that heterogeneity of presentation and response to treatment is underscored by important clinical and histopathological factors that should be considered in the workup and management of these conditions.
Keywords
Introduction
Kidney disease is common after stem cell transplant (SCT), with de novo glomerulopathies affecting 1–6% of recipients of allogeneic SCT. 1 Membranous nephropathy (MN) is the most commonly identified glomerulopathy, followed by minimal change nephropathy (MCN); focal segmental glomerulosclerosis (FSGS) and proliferative glomerulopathies occur at lower rates.1 –4 Weaning GvHD prophylaxis often precedes glomerulopathy manifestation; increasingly, some glomerulopathies are considered forms of kidney GvHD2,5 and a target antigen in post-SCT MN has recently been described. 2
We evaluated all kidney biopsies of allogeneic SCT recipients undertaken between 2019 and 2022 at Alfred Health, a major site for SCT in Victoria, Australia. We describe seven patients with nephrotic syndrome secondary to de novo glomerulopathy onset after allogeneic SCT. In the changing landscape of post-SCT glomerulopathy, our cases demonstrate that heterogeneity of presentation and response to treatment is underscored by important clinical and histopathological factors that should be considered in the workup and management of these conditions.
This case series report was prepared following CARE Guidelines. 6
Cases
Patient case summaries and kidney biopsy histopathology findings are displayed in Tables 1 and 2, respectively. Clinical outcomes are displayed in Table 3.
Demographics, clinical information and kidney biopsy diagnosis of included patients.
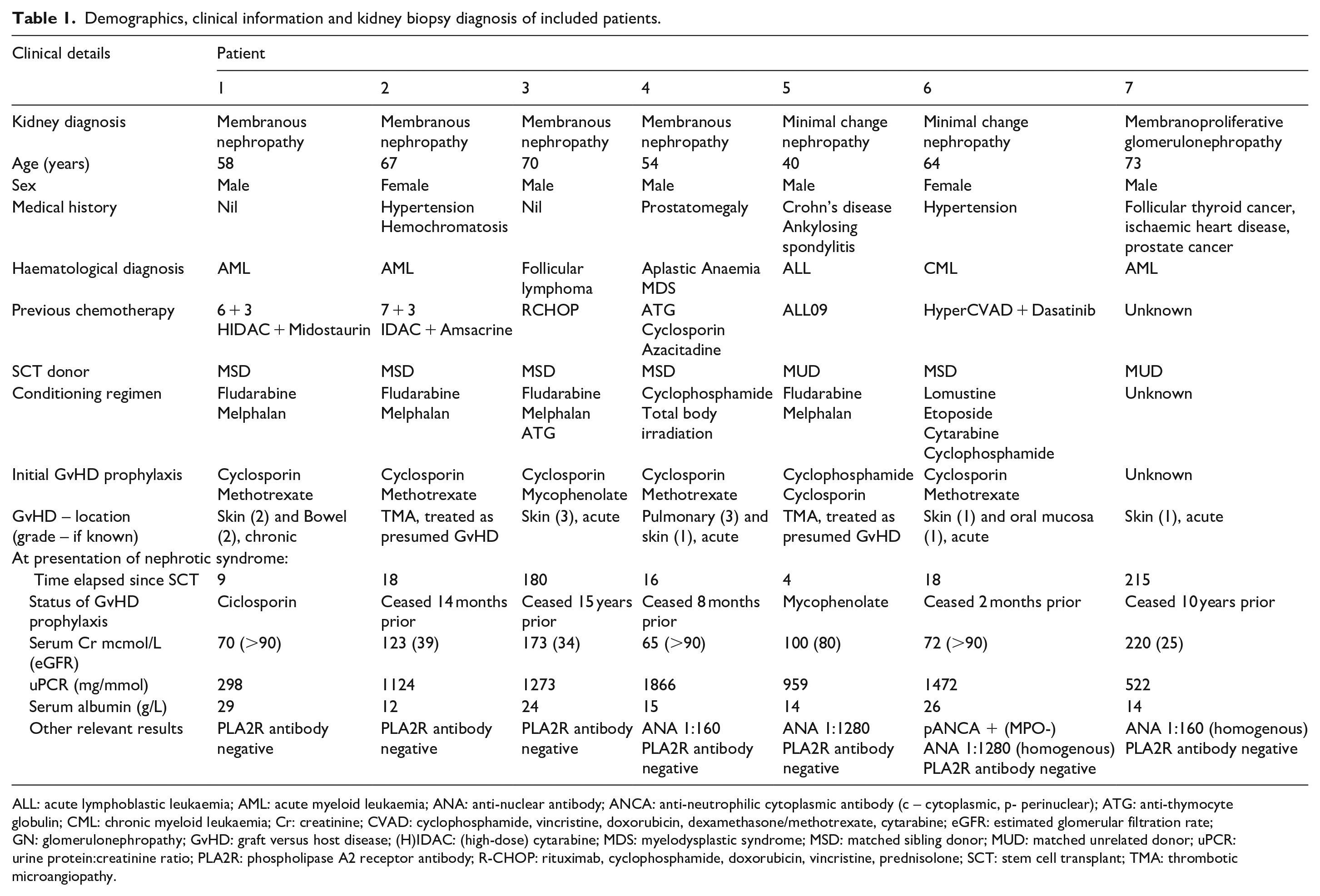
ALL: acute lymphoblastic leukaemia; AML: acute myeloid leukaemia; ANA: anti-nuclear antibody; ANCA: anti-neutrophilic cytoplasmic antibody (c – cytoplasmic, p- perinuclear); ATG: anti-thymocyte globulin; CML: chronic myeloid leukaemia; Cr: creatinine; CVAD: cyclophosphamide, vincristine, doxorubicin, dexamethasone/methotrexate, cytarabine; eGFR: estimated glomerular filtration rate; GN: glomerulonephropathy; GvHD: graft versus host disease; (H)IDAC: (high-dose) cytarabine; MDS: myelodysplastic syndrome; MSD: matched sibling donor; MUD: matched unrelated donor; uPCR: urine protein:creatinine ratio; PLA2R: phospholipase A2 receptor antibody; R-CHOP: rituximab, cyclophosphamide, doxorubicin, vincristine, prednisolone; SCT: stem cell transplant; TMA: thrombotic microangiopathy.
Summary of histopathological findings from kidney biopsy.
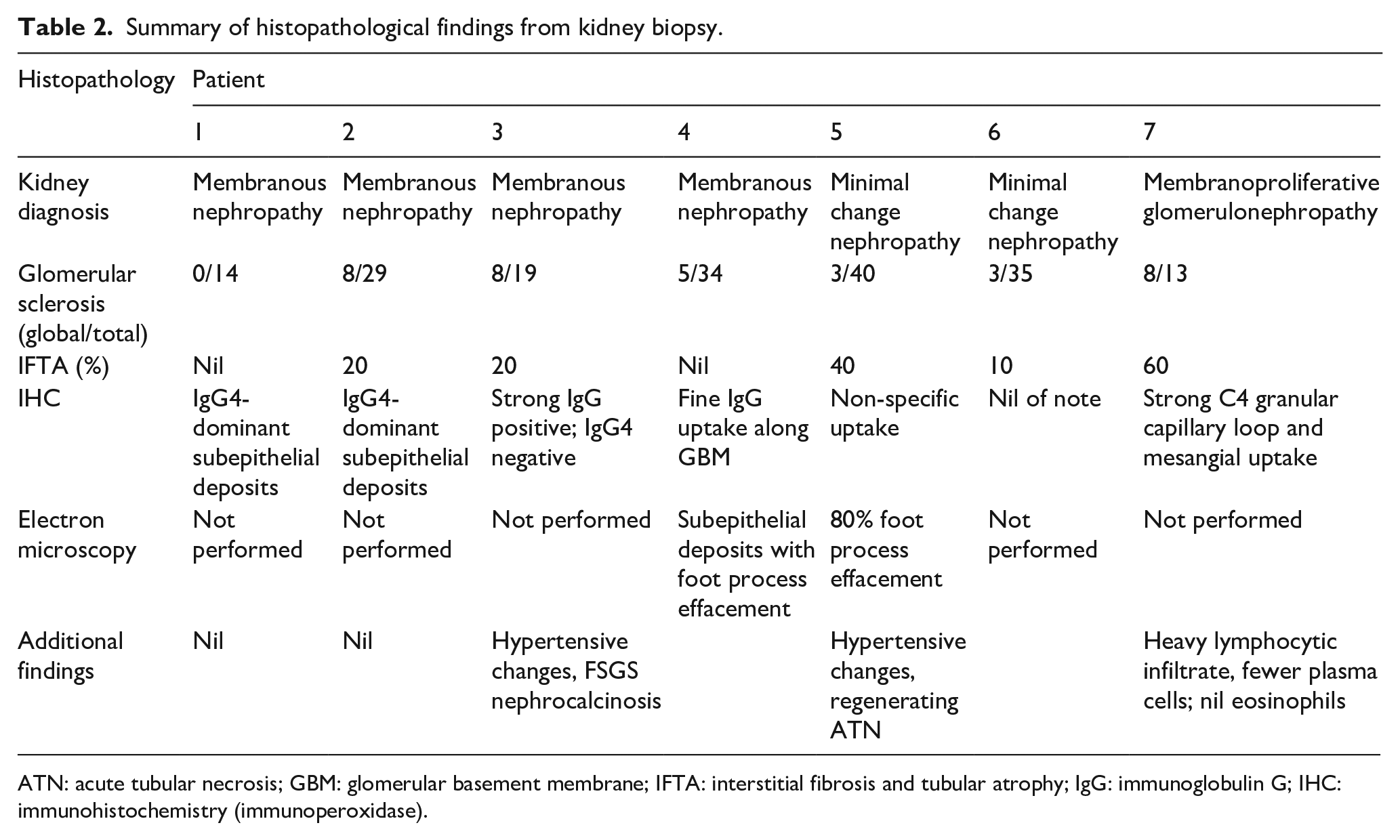
ATN: acute tubular necrosis; GBM: glomerular basement membrane; IFTA: interstitial fibrosis and tubular atrophy; IgG: immunoglobulin G; IHC: immunohistochemistry (immunoperoxidase).
Patient treatment and response.
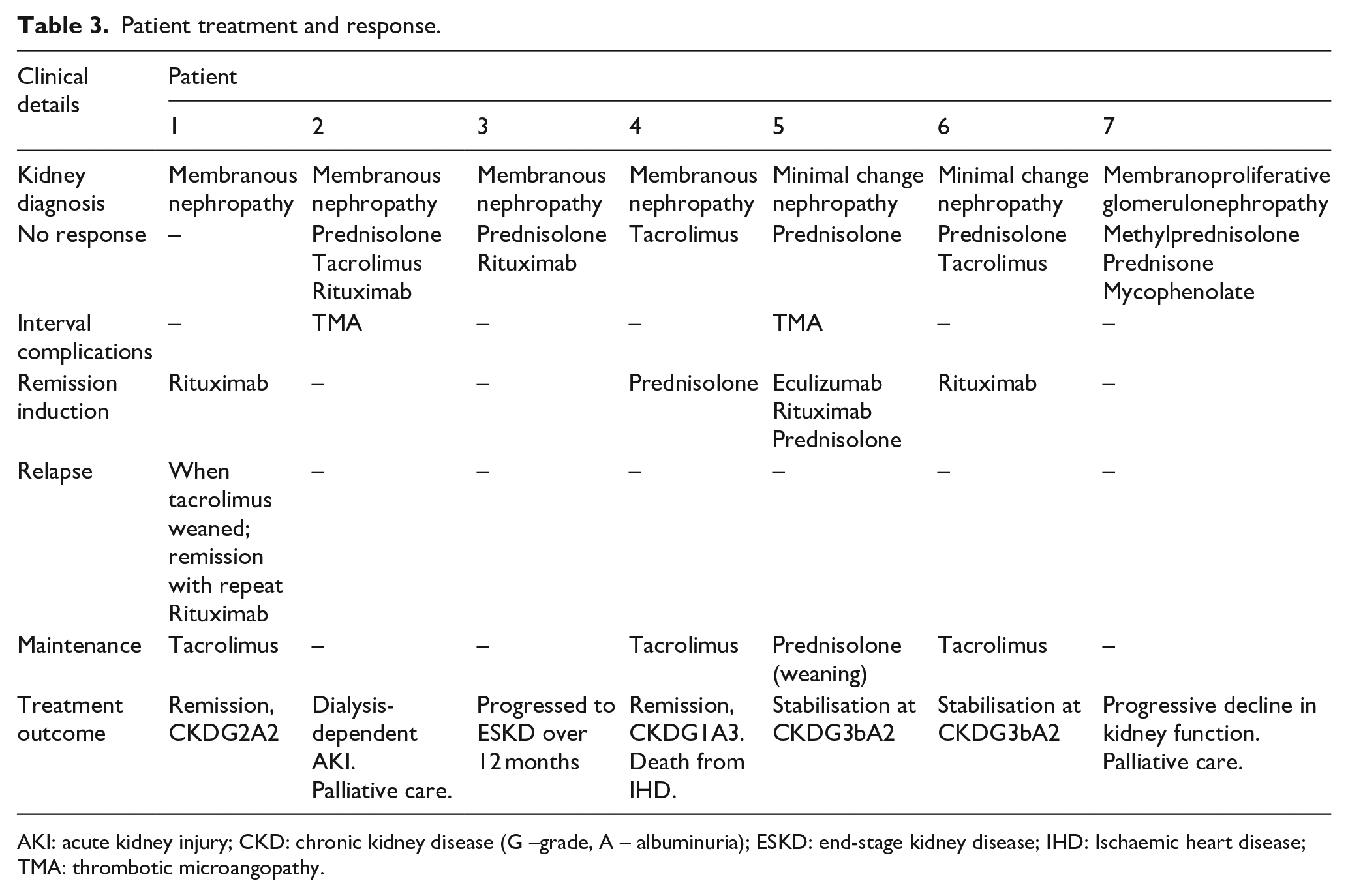
AKI: acute kidney injury; CKD: chronic kidney disease (G –grade, A – albuminuria); ESKD: end-stage kidney disease; IHD: Ischaemic heart disease; TMA: thrombotic microangopathy.
Patient 1
A white male in his fifties with no significant medical history was diagnosed with acute myeloid leukaemia (AML). After 7 + 3 cytarabine/anthracycline induction chemotherapy and high-dose cytarabine (HIDAC) and midostaurin consolidation, he received a fludarabine-melphalan conditioned matched sibling donor (MSD) SCT. GvHD prophylaxis was with cyclosporin and methotrexate; he later developed Grade 2 chronic GvHD of skin and bowel.
Nine months after SCT, he presented with nephrotic syndrome, urine protein 3.8 g/24 h, serum albumin 29 g/L. Urine microscopy demonstrated 5 × 106/L erythrocytes, non-dysmorphic. Kidney function was preserved, serum creatinine 70 mcmol/L (eGFR >90 mL/min/1.73 m2 total body surface area (TBSA)). Glomerulonephritis (GN) screen, including anti-Phospholipase A1 receptor (PLA2R) antibody, was negative.
Pathology findings
Kidney biopsy (Figure 1, 1(a–c)) demonstrated MN with no interstitial fibrosis or inflammation. Glomeruli were viable, of normal cellularity, with patent capillary loops. Jones silver stain demonstrated smooth linear glomerular basement membrane (GBM) without spiking. Masson trichrome stain demonstrated fine fuscinophilic subepithelial deposits without subendothelial or mesangial deposits. Immunoperoxidase stains demonstrated moderate to strong (2+ to 3+) glomerular basement membrane (GBM) IgA, IgG, IgM and C3 and weak (1+) C4 and C1q staining. There was no mesangial staining. There was strong (3+) IgG4 GBM staining. Electron microscopy (EM) was not undertaken.
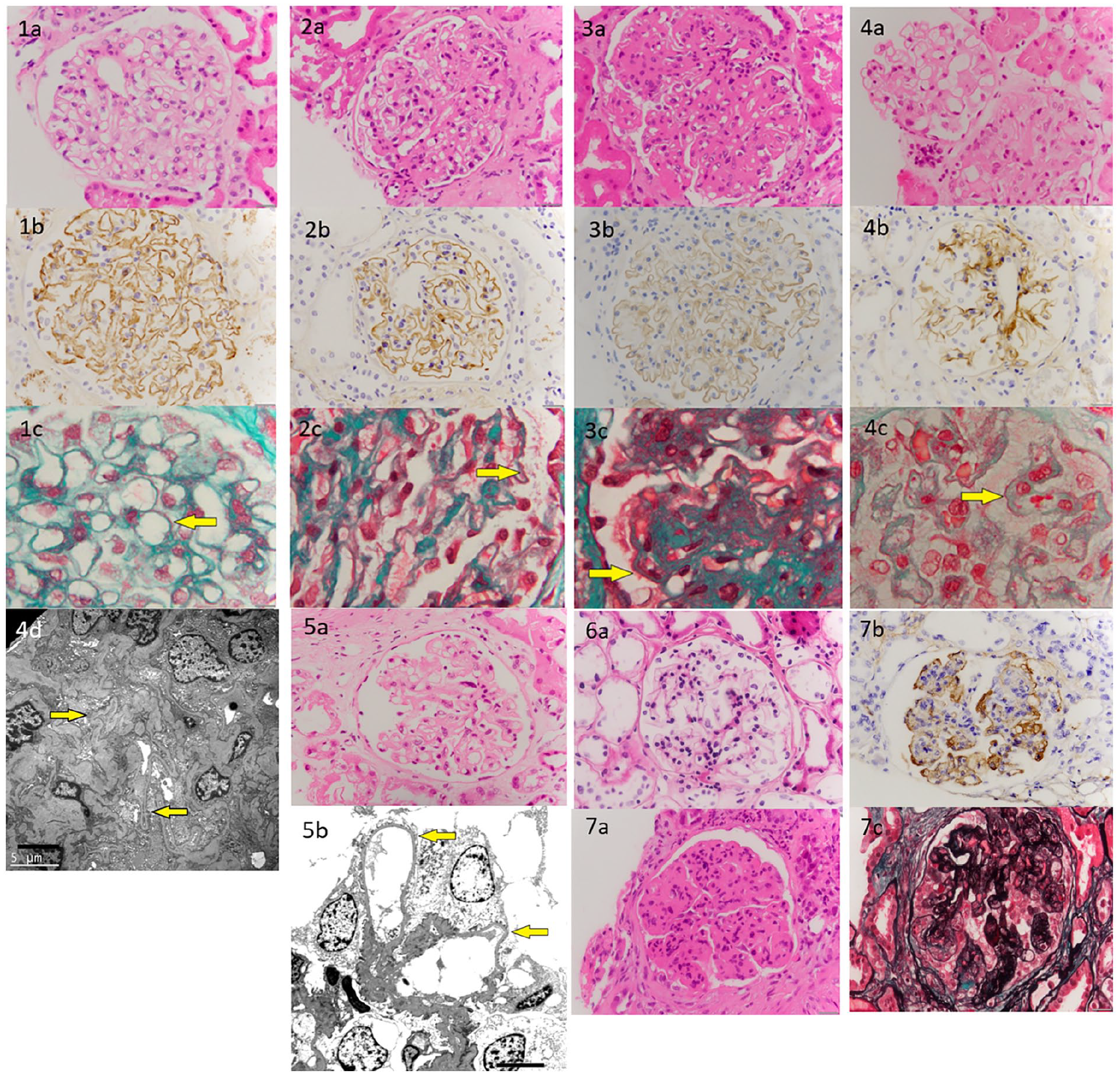
1. Kidney biopsy under light microscopy: (a) haematoxylin and eosin × 400 magnification demonstrating viable glomeruli with no interstitial fibrosis or inflammation, (b) IgG immunoperoxidase × 400 magnification demonstrating moderate to strong (2+ to 3+) glomerular basement membrane (GBM) staining, (c) Masson trichrome × 1000 magnification demonstrating fine fuscinophilic subepithelial deposits (yellow arrow) without subendothelial or mesangial deposits. 2. Kidney biopsy under light microscopy: (a) haematoxylin and eosin × 400 magnification demonstrating mild thickening of the GBM with mild mesangial matrix expansion, (b) IgG4 immunoperoxidase × 400 magnification with strong (3+) GBM, (c) Masson trichrome × 1000 magnification demonstrating fine fuscinophilic subepithelial deposits (yellow arrow). 3. Kidney biopsy under light microscopy: (a) haematoxylin and eosin × 400 magnification demonstrating increased mesangial matrix and foci of segmental sclerosis, (b) IgG Immunoperoxidase × 400 magnification demonstrating strong (3+) granular GBM staining for IgG only (c) Masson trichrome × 1000 magnification demonstrating very faint granular fuscinophilic deposits along capillary basement membranes (yellow arrow). 4. Kidney biopsy under light microscopy: (a) haematoxylin and eosin × 400 magnification demonstrating a normally formed glomerulus with mild focal increase in mesangial cellularity, (b) IgG Immunoperoxidase × 400 magnification demonstrating fine granular staining along the GBM, (c) Masson trichrome × 1000 magnification demonstrating faint granular fuscinophilic deposits along capillary basement membranes (yellow arrow), and (d) electron microscopy demonstrating subepithelial deposits with foot process effacement (yellow arrows). 5. Kidney biopsy under light microscopy: (a) haematoxylin and eosin × 400 magnification demonstrating some glomerular matrix expansion with GBM thickening and (b) electron microscopy magnification demonstrating 80% foot process effacement (yellow arrows). 6. Kidney biopsy under light microscopy: (a) haematoxylin and eosin × 400 magnification demonstrating normal appearance of the glomerulus. 7. Kidney biopsy under light microscopy: (a) haematoxylin and eosin × 400 magnification demonstrating increased mesangial matrix and accentuated glomerular lobularity, (b) Masson trichome × 400 magnification demonstrating occasional subendothelial fuscinophilic deposits, along with fine mesangial deposits (c) Silver stain × 400 magnification demonstrating mesangial matrix increase and diffuse double contouring of the GBM.
Clinical follow-up
Remission was achieved with two doses of rituximab 1 g given 2 weeks apart, and oral prednisolone 1 mg/kg. Prednisolone was weaned and ceased over 1 month and changed to tacrolimus (T0 target 6–8 ng/L) for maintenance. Tacrolimus was slowly weaned over 12 months; he experienced a relapse with rising proteinuria. Treatment with two doses of rituximab 1 g given 2 weeks apart and recommencing tacrolimus restored complete remission which is currently maintained.
Patient 2
A white female in her 60s with haemochromatosis and medication-controlled hypertension was diagnosed with AML, initially treated with 7 + 3 cytarabine/anthracycline induction chemotherapy with remission, then intermediate-dose cytarabine/amsacrine before fludarabine-melphalan conditioned MSD SCT. GvHD prophylaxis was with cyclosporin and methotrexate; she did not develop GvHD.
Eighteen months post SCT, she presented with nephrotic syndrome, spot urine protein:creatinine ratio 1124 mg/mmol, serum albumin 12 g/L and acute kidney injury, serum creatinine 123 mcmol/L from baseline 78 mcmol/L. GN screen, including PLA2R antibody, was negative.
Pathology findings
Kidney biopsy (Figure 1, 2(a–c)) demonstrated early MN with mild interstitial fibrosis. Glomeruli demonstrated 28% global sclerosis. Viable glomeruli demonstrated mild GBM thickening with mild mesangial matrix expansion. Silver stain demonstrated predominantly smooth GBM without obvious spiking; fine fuscinophilic subepithelial deposits were seen on trichrome stain. Immunoperoxidase stains demonstrated strong (3+) IgG (predominantly IgG4) GBM and moderate (2+) IgA, IgM and C3 staining without C4 or C1q staining.
Clinical follow-up
The patient was treated with two doses of rituximab 1 g given 2 weeks apart, oral prednisolone 1 mg/kg and tacrolimus (T0 target 6–8 ng/L). She subsequently developed thrombotic microangiopathy (TMA). Serum C3 and C4 were low-normal; C5b-9 and CH50 testing were not available at our centre. Tacrolimus was ceased, she received eculizumab and was changed to sirolimus and myocophenolate mofetil for presumed GvHD; proteinuria worsened, rising to spot urine protein:creatinine ratio of 4362 mg/mmol with progressive kidney impairment. Serum C3 and C4 were not re-tested prior to her transition to palliative care and death during acute admission.
Patient 3
A white male in his 50s with no significant medical history was diagnosed with non-Hodgkin lymphoma. After initial treatment with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisolone) without response, he received a fludarabine-melphalan and anti-thymocyte globulin conditioned MSD SCT. GvHD prophylaxis was with ciclosporin and mycophenolate mofetil; he later developed Grade 3 acute skin GvHD. He experienced disease relapse 4 years later, treated with donor lymphocyte infusion.
Fifteen years after SCT, whilst in remission from his original non-Hodgkin lymphoma, he presented with nephrotic syndrome, urine protein 6 g/24 h, serum albumin 24 g/L and kidney impairment, serum creatinine rose from 86 to 173 mcmol/L over 12 months (eGFR 80–34 mL/min/ 1.73 m2 TBSA). GN screen, including PLA2R antibody, was negative.
Pathology findings
Kidney biopsy (Figure 1, 3(a–c)) demonstrated MN with focal segmental glomerulosclerosis, mild interstitial fibrosis and atrophy, nephrocalcinosis and hypertensive vasculopathy. Glomeruli were 42% globally sclerosed; viable glomeruli demonstrated increased mesangial matrix and focal segmental sclerosis. Silver stain revealed GBM thickening with subepithelial spiking. Immunoperoxidase stains demonstrated strong (3+) granular IgG GBM staining (IgG4 was negative), with weak (1+) GBM staining for IgA, IgM, C1q (focal) and C3.
Clinical follow-up
The patient was treated with oral prednisolone 1 mg/kg and two doses of Rituximab 1 g given 2 weeks apart, without response. Over the next 12 months, he progressed to end-stage kidney disease (ESKD) with ongoing nephrotic range proteinuria. He was commenced on peritoneal dialysis.
Patient 4
A white male in his fifties with benign prostatic hypertrophy and prior aplastic anaemia treated with antithymocyte globulin and cyclosporin 15 years prior developed high-risk myelodysplastic syndrome (MDS). He was treated with Azacitadine ± eltrombopag 7 and underwent MSD SCT after myeloablative conditioning with cyclophosphamide and total body irradiation. GvHD prophylaxis was with cyclosporin and methotrexate; he developed severe acute pulmonary GvHD and mild skin GvHD.
Sixteen months post SCT, he presented with nephrotic syndrome, spot urine protein:creatinine ratio 1866 mg/mmol and serum albumin 15 g/L. Kidney function was preserved, serum creatinine 65 mcmol/L (eGFR >90 mL/min/1.73 m2 TBSA). GN screen demonstrated positive anti-nuclear antibodies (ANA) at 1:160 titre (nucleolar pattern) and indeterminant p (perinuclear) and c (cytoplasmic) anti-nuclear cytoplasmic antibodies; myeloperoxidase (MPO) and anti-proteinase-3 (PR3) antibodies were negative. PLA2R antibody was negative.
Pathology findings
Kidney biopsy (Figure 1, 4(a–d)) demonstrated MN, with mild interstitial inflammation and fibrosis. Glomeruli demonstrated 15% global sclerosis. Patchy interstitial inflammation, comprising lymphocytes and plasma cells, involved 20% of the cortical parenchyma. Viable glomeruli were normally formed with mild focally increased mesangial cellularity. There was mild GBM thickening, trichrome stain showed very faint granular fuscinophilic deposits along capillary basement membranes with immunoperoxidase demonstrating fine granular staining for IgG, IgM and C1q. EM demonstrated subepithelial deposits.
Clinical follow-up
The patient was initially treated with tacrolimus (T0 target 6–8 ng/L); corticosteroids were omitted due to concomitant pulmonary aspergillosis. After 1 month without response, oral prednisolone 1 mg/kg was added, and remission was achieved. He was transitioned to maintenance tacrolimus after 2 months and remission was maintained. Kidney function was preserved throughout. He died of ischaemic heart disease 4 years later.
Patient 5
A white male in his forties with Crohn’s disease and Ankylosing Spondylitis treated with Adalimumab was diagnosed with acute lymphoblastic leukaemia and treated with induction prednisolone, methotrextate, danorubicin, vincristine, pegaspargase with Blinatumomab consolidation (ALLG ALL09 SuBliME trial) 8 followed by reduced-intensity fludarabine-melphalan conditioned matched unrelated donor (MUD) SCT. GvHD prophylaxis was with cyclophosphamide and cyclosporin, later transitioned to mycophenolate mofetil. He did not develop GvHD.
From a baseline pre-SCT serum creatinine of 65 mcmol/L (eGFR >90 mL/min/1.73 m2 TBSA), he had multiple acute kidney insults related to supratherapeutic ciclosporin levels and daptomycin-related rhabdomylosis. New baseline serum creatinine 3 months after SCT was 120 mcmol/L (eGFR 65 mL/min/1.73 m2 TBSA). One month later, he developed nephrotic syndrome with spot urine protein:creatinine ratio 967 mg/mmol and serum albumin 14 g/L. ANA was positive at 1:1280 titre (homogenous pattern); extractable nuclear antibodies were negative. GN screen, including PLA2R antibody, was otherwise negative.
Pathology findings
Kidney biopsy (Figure 1, 5(a and b)) demonstrated regenerating ATN, moderate background interstitial fibrosis with hypertensive vasculopathy. There was glomerular matrix expansion with GBM thickening. Immunoperoxidase demonstrated non-specific immunoglobulin staining, particularly IgM. EM demonstrated 80% foot process effacement, in keeping with MCN.
Clinical follow-up
The patient was commenced on oral prednisolone 1 mg/kg but developed TMA and acute kidney injury 2 months later, serum creatinine rose to 320 mcmol/L. Serum C3 and C4 were low-normal; C5b-9 and CH50 testing were not available at our centre. He was treated with eculizumab and rituximab weekly for presumed GvHD. There was no change in his serum C3 or C4 levels after treatment with eculizumab. New baseline kidney function is serum creatinine 220 mcmol/L, eGFR 31 mL/min/1.73 m2 TBSA. Rituximab continued for 1 month and eculizumab for 5 months and prednisolone weaning continues.
Patient 6
A white female in her sixties with medication-controlled hypertension was diagnosed with chronic myeloid leukaemia (CML) which underwent lymphoblastic transformation and lymphoid blast crisis 2 years later. After induction chemotherapy with hyper-CVAD (cyclophosphamide, vincristine, doxorubicin, dexamethasone/methotrexate, cytarabine), she received a myeloablative LACE (lomustine, cytarabine, cyclophosphamide, etoposide) conditioned MSD SCT. GvHD prophylaxis was with cyclosporin and methotrexate; she developed acute skin and oral mucosal GvHD.
Twelve months later, her sibling donor developed nephrotic syndrome secondary to biopsy-proven MCN. His steroid-resistant disease was successfully treated with cyclophosphamide, remission is maintained with tacrolimus (T0 target 5–7 ng/L).
Six months later (18 months after SCT), the SCT recipient presented with nephrotic syndrome with spot urine protein:creatinine ratio 1472 mg/mmol and serum albumin 26 g/L. Urine microscopy identified 99 × 106/L erythrocytes; non-dysmorphic. Kidney function was preserved, serum creatinine was 72 mcmol/L (eGFR >90 mL/min/1.73 m2 TBSA). GN screen demonstrated positive p-ANCA; MPO and PR3 antibodies were negative. PLA2R antibody was negative. ANA was positive, 1:1280 titre (homogenous pattern).
Pathology findings
Kidney biopsy (Figure 1, 6(a)) demonstrated hypertensive vasculopathy with 10% global glomerular sclerosis; no specific glomerular pathology was identified on light microscopy or immunoperoxidase staining. Sample size was insufficient for EM. Note, the sibling donor’s kidney biopsy demonstrated weak linear IgG uptake of capillary loops and GBM on immunofluorescence and EM demonstrated 90% foot process effacement, consistent with MCN.
Clinical follow-up
The patient was treated initially with oral prednisolone 1 mg/kg for presumed minimal change nephropathy, with minimal response. Adding tacrolimus (T0 target 6–8 ng/L) achieved partial response; proteinuria remained nephrotic range. Addition of two doses of rituximab 1 g given 2 weeks apart achieved improvement in proteinuria with spot urine protein:creatinine ratio 35 mg/mmol and serum albumin 40 g/L; kidney function declined over the following 3 years to a new baseline serum creatinine of 128 mcmol/L (eGFR 37 mL/min/1.73 m2 TBSA).
Patient 7
A white male in his fifties was diagnosed with AML treated with MUD SCT. Details for induction chemotherapy, conditioning regimen and GvHD prophylaxis were not available from the external health service; he was known to have developed mild chronic skin GvHD. In the following 18 years, he developed ischaemic heart disease, local prostate cancer treated with trans-urethral resection, and metastatic follicular thyroid cancer refractory to radioactive iodine, treated with levantanib.
During an inpatient admission for pneumonia and acute myocardial infarction, he developed acute on chronic kidney injury, serum creatinine rose from 145 to 215 mcmol/L. He had nephrotic-range proteinuria with spot urine protein:creatinine ratio 552 mg/mmol and serum albumin 14 g/L. GN screen demonstrated positive ANA, 1:160 titre (homogenous pattern) but was otherwise negative, including PLA2R antibody, urine and serum protein electrophoresis and free light chains.
Pathology findings
Kidney biopsy (Figure 1, 7(a–c)) demonstrated features of immune complex membranoproliferative glomerulonephritis (MPGN), with advanced global glomerulosclerosis (62%), and severe fibrosis. Of viable glomeruli, all demonstrated increased mesangial matrix, accentuated glomerular lobularity and most had segmental sclerosis. Silver stain confirmed mesangial matrix increase and diffuse GBM double contouring. Trichrome stain demonstrated occasional subendothelial fuscinophilic deposits with fine mesangial deposits. Immunoperoxidase demonstrated strong (3+) granular capillary loop and mesangial staining for IgM and C4, with weak (1+) staining for C1q, IgA, IgG and C3. Kappa and lambda light chain immunoperoxidase stains were negative. There was interstitial fibrosis and tubular atrophy of 60% of the cortex and a moderately heavy interstitial infiltrate of lymphocytes, with smaller numbers of plasma cells and occasional neutrophils, but no eosinophils.
Clinical follow-up
The patient was treated with pulse methylprednisolone 250 mg IV then oral prednisolone 1 mg/kg, and mycophenolate mofetil, without response. During admission, he was diagnosed with a gastro-intestinal neuroendocrine tumour. He requested cessation of active management and died 12 months later.
Discussion
De novo glomerulopathies complicate 1–6% of allogeneic SCT, 1 with nephrotic syndrome reported in <1% of allogeneic SCT recipients. 9 MN is the most commonly identified pathology, followed by MCN,1 –4 similar in our series. Patient factors, underlying haematological disease and treatment-related risk factors remain poorly understood. 10 We describe a diverse patient cohort of nephrotic syndrome after allogeneic SCT, and discuss important factors that should be considered in the workup and management of these conditions, including interval between SCT and nephrotic syndrome, concomitant GvHD and immunosuppression weaning, and underlying kidney histopathology.
Time course and pathophysiology
Most nephrotic syndrome after SCT demonstrates a temporal relationship to immune reconstitution and withdrawal of immunosuppressive GvHD prophlyaxis. 1 In our case series, median time from SCT to nephrotic syndrome was 16 months, similar to the wider literature 4 ; there was significant variability, from 4 months to 18 years. Patient 1 developed nephrotic syndrome whilst receiving GvHD prophylaxis, as occurs in up to 40% of patients 4 ; the majority of patients in our series developed nephrotic syndrome months after withdrawal of GvHD prophylaxis (median 9 months). Many post-SCT glomerulopathies, and MN in particular, are described as a form of ‘kidney specific’ GvHD mediated by allo-reactive donor T-cells against recipient tissues 11 and a systematic review of 116 cases of post-SCT nephrotic syndrome (1988–2015) reported that nephrotic syndrome onset often occurs concomitantly with acute or chronic GvHD, with temporal relationship to weaning or cessation of GvHD prophylaxis. 4
De novo glomerulopathies also occur after autologous SCT, for which GvHD cannot be causative. Other factors are therefore implicated, such as immune dysregulation from SCT conditioning regimens and impaired immune tolerance of reconstituted B-cells after myeloablative conditioning.12,13 The pathogenesis of late-onset glomerulopathy after SCT may similarly differ from that occurring in the acute post-allogeneic SCT period. Two patients (Patients 3 and 7) developed nephrotic syndrome many years after SCT, with histopathological diagnoses of MN and MPGN, respectively. In these settings, de novo glomerulopathy may be related to conditions other than GvHD, such as Patient 7’s newly diagnosed gastro-intestinal neuroendocrine tumour or pre-existing metastatic thyroid cancer, as has been described in the literature.14,15
Histopathology
Histopathology of post-SCT MN is diverse. Changes typical of primary MN are often seen, with IgG4-dominant complexes localized to the sub-epithelium; atypical histopathology is also observed. 16 Even among patients with typical primary MN histopathology, PLA2R antibodies are usually negative in post-SCT MN. 17 In our case series, both typical and atypical histology for primary MN was observed, all four patients with MN demonstrated subepithelial patterns of IgG uptake; only two were IgG4-dominant and all were PLA2-R antibody negative. Recent work has identified novel protein Protocadherin FAT1 in podocyte intercellular junctions as the target antigen in the majority of cases of SCT-associated MN, with staining pattern typical of primary MN, 2 which may support the ‘kidney specific’ GvHD post-SCT MN hypothesis.
Up to 20% of post-SCT nephrotic syndrome is attributable to MCN. 4 Previously described, Patient 6 is a unique case wherein nephrotic syndrome developed shortly after the same in her sibling donor. 18 Recent work demonstrated circulating anti-Nephrin autoantibodies in sera co-localised with IgG in GBM of 1/3rd of a biopsy-proven MCN patient cohort, 19 which may be analogous to PLA2R antibodies in MN in the general population and protocadherin FAT-1 in post-SCT MN patient populations. This is hypothesised to account for previously-described delicate punctate IgG staining identified in MCD patient subsets. 19 With anti-Nephrin autoantibody testing only recently described, these tests were not undertaken in our patients; Patient 6’s SCT donor kidney biopsy demonstrated weak IgG positive GBM immunofluorescence, similar to that seen in anti-Nephrin autoantibody-positive patients in the aforementioned cohort. 19 This is a hypothesis-generating case that MCN pathogenesis may have been related to a SCT-transmitted circulating factor or donor-derived memory B-cells.
Treatment
Treatment choices for post-SCT glomerulopathy have largely been adopted from that used for idiopathic nephrotic syndrome in the non-SCT setting 10 ; the extent to which these populations are analogous is unclear. Most post-SCT MN appears responsive anti-CD20 therapy, 20 which has also been successfully deployed to prophylactically reduce chronic GvHD, 21 and induce remission in MCN. 22 There remains however, significant heterogeneity in treatment approaches to post-SCT nephrotic syndrome, 4 unsurprising considering the diversity of underlying pathologies. It is important to consider whether nephrotic syndrome is likely related to unrelated to SCT when considering treatment options, time-course from SCT to nephrotic syndrome and patient comorbidities can help inform this consideration.
In our case series, Rituximab was used to treat Patient 1’s original MN and later relapse, and Patients 5 and 6’s MCN; the former with intercurrent treatment for TMA. Patient 4 achieved remission with calcineurin-inhibitor and steroids, preferentially avoiding longer-acting immunosuppression given his concomitant pulmonary aspergillosis. Patients 2 and 3 had MN on kidney biopsy but did not respond to Rituximab; Patient 2 developed TMA and died due to related complications. Patients 3 and 7 had long interval periods between SCT and developing nephrotic syndrome; their cases may represent disease unrelated to the original SCT and/or GvHD. Poor response to treatment of late-onset nephrotic syndrome is reported in the literature; late-onset nephrotic syndrome is less responsive to the high (>80%) rates of complete remission described for patients with relatively more acute-onset post-SCT MCN and MN (27–59%). 4 >15 years elapsed between Patient 3’s SCT and nephrotic syndrome onset; his non-response to Rituximab and progressive kidney impairment may be attributable to the dual pathology of MN and FSGS. Although published literature regarding post-SCT MPGN is onset scarce, 10 up to 5% of post-SCT nephrotic syndrome may be attributable to MPGN. 4 For Patient 7, the almost two-decade interval between SCT and nephrotic syndrome, and comorbid metastatic cancer, may similarly suggest that the underlying pathophysiology of his late-onset nephrotic syndrome differs from the previously-described SCT-related immune causes. It may be considered therefore, that anti-CD20 therapy effectively treated most patients where nephrotic syndrome onset temporally related to SCT and/or GvHD prophylaxis weaning, where comorbidities did not render it unsuitable for use.
Conclusion
This case series describes seven patients with nephrotic syndrome onset after allogeneic SCT, with diverse indications for SCT, kidney histopathology and subsequent treatment. Time post SCT, concomitant GvHD and underlying comorbidities were relevant considerations in their clinical courses, likely underscoring heterogeneity of presentation and responses to treatment. Future improvements in delineating pathophysiology of underlying disease entities are required to tailor treatments accordingly and improve patient outcomes.
Footnotes
Acknowledgements
We gratefully acknowledge Dr Sophia Otto, Director – SA Pathology and Dr Deborah Gomes, Anatomical Pathology Registrar – SA Pathology, and John Brealey, Senior electron microscopy scientist – SA Pathology, for their assistance with some of the cases included in this series.
Availability of data
The data that support this case report are available from Alfred Health, but restrictions apply to the availability of these data due to patient confidentiality. Data are however available from the authors upon reasonable request, with permission of included patients and the Alfred Health Ethics Board.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval and consent to participate
Institutional ethics was provided by Alfred Health (Project number: 498/22). Informed signed consent to participate was obtained for all living patients as per Alfred Health guidelines, included in patient electronic medical record. Consent waiver for deceased patients was granted by Alfred Health Ethics review board.
Consent to publish
Informed signed consent to publish was obtained for all living patients as per Alfred Health guidelines, included in patient electronic medical record. Consent waiver for deceased patients was granted by Alfred Health Ethics review board (project number 498/22).
Trial registration
NA
Guarantor
A/Prof Gopal Basu
Authorship
DF, VW, SP and BG undertook case selection. DF wrote the main manuscript. VW, SP and BG contributed to editing and review. AP performed the histopathological examination of the recipient kidney and photographed images for inclusion. All authors read and approved the final manuscript.