
Abstract
Sarcoid-Like Reaction is a systemic inflammatory granulomatous condition that does not meet the criteria for systemic Sarcoidosis—it is triggered by a known underlying factor, such as immunosuppression. Here we report a case of acute interstitial nephritis without granuloma in the context of a Sarcoid-Like systemic disease in the three-year period after an autologous stem cell transplant. The patient presented with a suspected relapse, associated with hemolytic anemia and an increase of serum creatinine. The investigation showed non-caseating epithelioid cell granuloma in a chest lymph node and in bone marrow, associated with combined hypogammaglobulinemia. Evaluation of the kidney tissue demonstrated acute interstitial nephritis.
Keywords
Introduction
Acute interstitial nephritis (AIN) is a kidney lesion characterized by the presence of inflammatory infiltrates and edema within the interstitium, usually associated with an acute increase in serum creatinine. A significant proportion of AIN has an oligosymptomatic presentation and drug-induced AIN is the most common etiology (70–75% of the cases). Autoimmune disorders or other systemic diseases such as systemic lupus erythematosus, Sjögren’s disease, and sarcoidosis represents 10–20% of the cases.1,2
Sarcoidosis is a multisystem inflammatory granulomatous disorder of unknown etiology that affects individuals worldwide and is characterized pathologically by the presence of noncaseating granulomas in involved organs. It typically presents with one or more of the following abnormalities: bilateral hilar adenopathy; pulmonary reticular opacities; skin, joint, and/or eye lesions. 3 Kidney involvement in sarcoidosis is a rare condition. 4 Tubulointerstitial nephritis is an uncommon manifestation and can be presented with or without granuloma formation.
When a patient develops non-infectious and non-caseating epithelioid cell granulomas triggered by a known underlying factor, that is, it does not meet the criteria for the diagnosis of systemic sarcoidosis, this entity is called a Sarcoid-Like Reaction (SLR). Sarcoid-Like (SL) granulomas have been increasingly described and most frequently associated with immunodeficiency, exposure to anti-TNF or checkpoint inhibitors, and several malignancies. 5 These cases typically present with intrathoracic involvement—pulmonary and/or mediastinal lymphadenopathy, but extrathoracic disease in liver, spleen, skin, and eye have also been reported. 3
We hereby report a case of AIN secondary to a SLR, associated with humoral immunodeficiency in the post-autologous stem cell transplant (ASCT) period of a patient treated for non-Hodgkin mantle cell lymphoma. To our knowledge, this is the first reported case of kidney sarcoid-like reaction after an ASCT.
Case report
A 69-year-old female patient was diagnosed with advanced Mantle cell Lymphoma in October 2015 and was treated with chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisone) between November 2015 and April 2016, followed by conditioning regimen BEAM (carmustine, etoposide, cytarabine, and melphalan) and a ASCT, achieving first complete remission in May 2016. The patient remained asymptomatic until November 2019, when a small movable lymph node measuring 1.5 cm was detected in the left lower neck/supraclavicular region. Positron Emission Tomography/Computed Tomography (PET-CT) evaluation showed several areas of abnormal uptake (left supraclavicular region, mediastinal, and hilar lymph nodes), with maximum SUV capture (4.0) by the mediastinal chain. This lymph node was biopsied through mediastinoscopy, revealing chronic granulomatous infiltrate without necrosis, which was negative for fungal and mycobacteria stains. As no other clinical manifestations were present, relapse was ruled out, and the patient remained under close surveillance.
In November 2020, she developed a Coombs-negative hypoproliferative hemolytic anemia, prompting a bone marrow biopsy, which demonstrated the same granulomatous findings of the first lymph node biopsy. At this point, serum creatinine (SCr) was 1.0 mg/dL (estimated glomerular filtration rate by the CKD-EPI equation: 61 mL/min/1.73 m2), and combined hypogammaglobulinemia was observed—serum IgA was 48.8 mg/dL (normal range 69–382 mg/dL) and serum IgG was 466 mg/dL (normal range 700–1600 mg/dL). In January 2021, prednisone 1 mg/kg/day was initiated due to worsening of anemia (Table 1), followed by a titration to 10 mg/day over the course of 6 months. In August 2021, coincident with weaning of corticosteroid, an increase in SCr over the course of several weeks was observed (peak 2.8 mg/dL) and the patient was referred to the Nephrology Clinic. Blood pressure was normal; no edema or other abnormal physical sign was observed. A full investigation, including urinalysis, complement analysis, serum, and urinary immunofixation, hepatitis and HIV serologies, and antineutrophil cytoplasmic antibody (ANCA), revealed no abnormalities, except for the presence of minor proteinuria (0.7 g/day). Renal echography and Doppler found no obstruction or stenosis.
Key findings through follow-up.
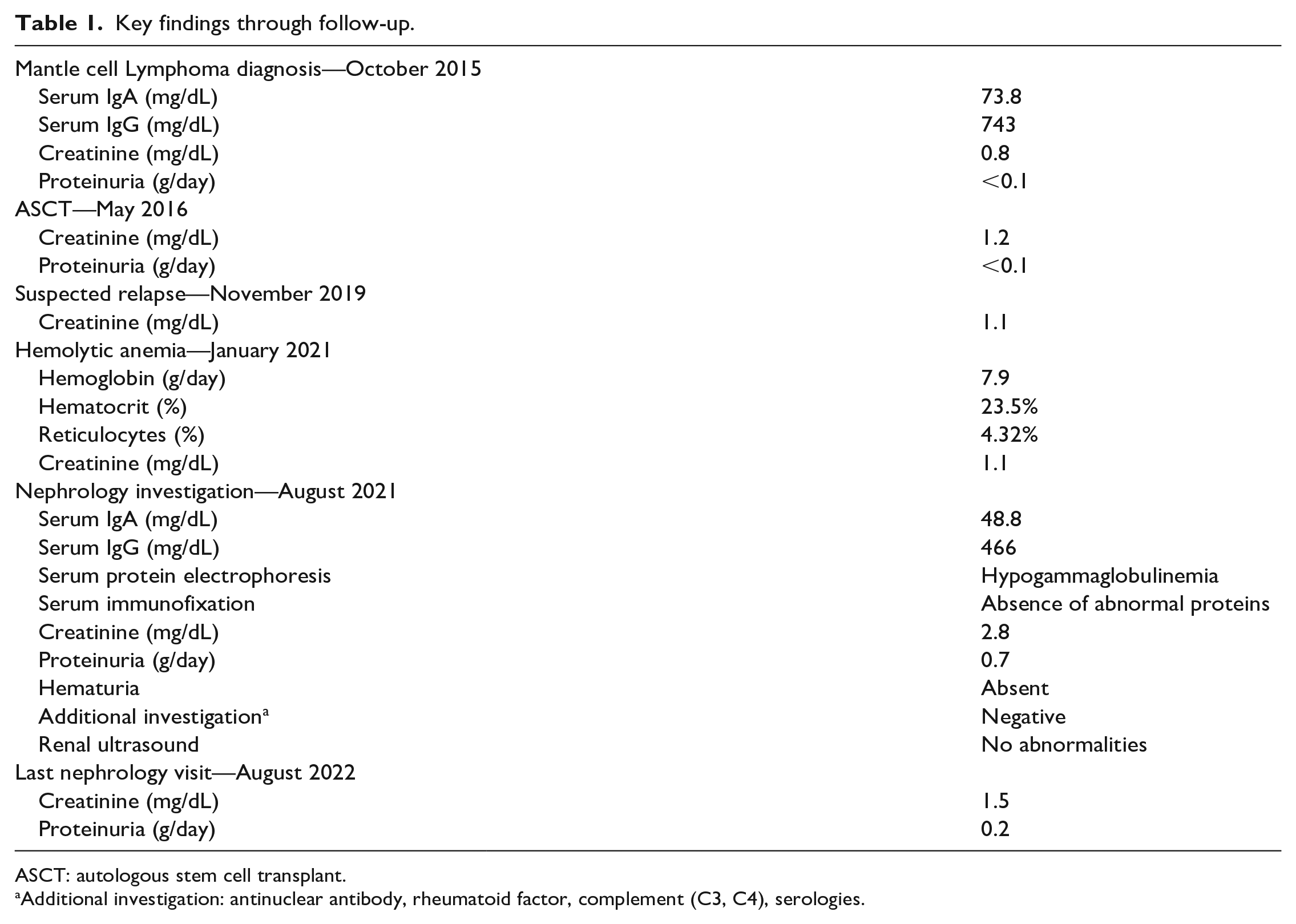
ASCT: autologous stem cell transplant.
Additional investigation: antinuclear antibody, rheumatoid factor, complement (C3, C4), serologies.
A percutaneous renal biopsy identified tubulitis and degenerative lesions of the epithelium with slightly thickened basal membrane; dissociated interstitium by edema, and infiltrate-granulomatous lesions, as all immunofluorescence stains were absent (Figure 1).
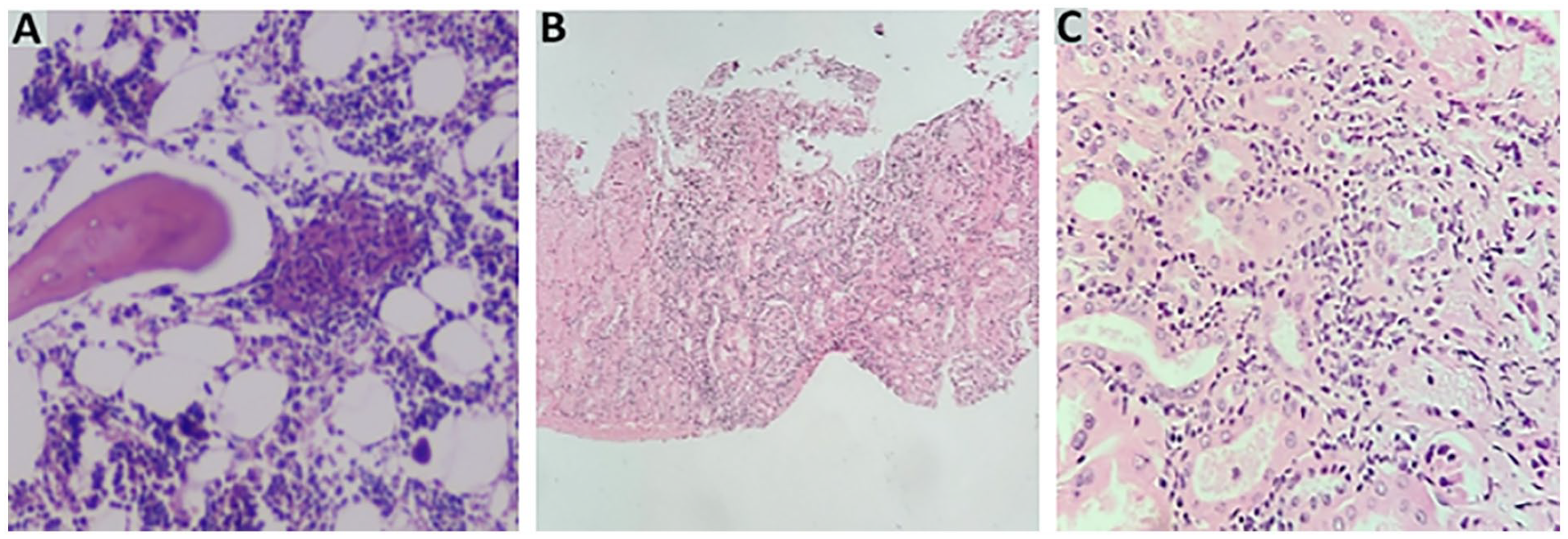
A) Bone Marrow biopsy with a non-caseating epithelioid cell granuloma in the intertrabecular space. B and C) Kidney biopsy demonstrating interstitial mononuclear cell infiltrate causing tubulitis.
After multidisciplinary discussion with Pathology, Hematology, Pulmonology, and Nephrology teams, a diagnosis of immunodeficiency-associated granulomatous disease or SLR post-ASCT was made. Prednisone dose was increased to 60 mg/day, with a decline of SCr to 2.0 mg/dL (eGFR 27 mL/min/1.73 m2) in the next two weeks. After two months, methotrexate 10 mg/week was initiated with the tapering of steroids. Ever since, the patient has been under nephrology care with a sustained SCr of 1.5 mg/dL (eGFR 37 mL/min/1.73 m2).
Discussion
Herein we reported a case of multiorgan-targeted SLR in the context of immunosuppression following an ASCT for a Mantle cell Lymphoma. There were histological sarcoid lesions in the lymph node and bone marrow, although no granuloma was found in the kidney. To our knowledge, this is the first reported case of AIN secondary to SLR after an ASCT.
Sarcoidosis is a disease of unknown origin, while SRL is considered to be secondary to an underlying condition, such as immunosuppression. The SLR is thought to occur due to antigens that trigger a T-cell-mediated local immune response. It is hypothesized that the participation of a dysregulated immune response in which a triggering antigen would promote dendritic cells to activate T-cell helper 1, resulting in massive cytokine production (IL-2, INF-gamma, and TNF-alfa), and mobilization of M1 macrophages to form the granuloma. 3 The development of SLR after SCT may derive from diverse immunologic derangements: immunosuppressive effect of chemotherapy or immunologic hypersensitivity to tumor antigenic factors, or even the consequence of an underlying immunologic disturbance. 6 In the post-SCT period, transitory immunodeficiency is expected until full immunologic reconstitution, which is mainly manifested as infectious diseases. In our case, the hypogammaglobulinemia may suggest a B-cell reconstitution deficiency, which should no longer be seen after 18 months of SCT.7,8 The relationship between common variable immunodeficiency and granulomatous disease is well established, and the hypogammaglobulinemia present in our patient could mimic this disorder and induce a SLR.
Granulomatous interstitial nephritis (GIN) secondary to sarcoidosis is a cause of kidney impairment and is present in 7–27% of all patients in autopsy studies. 9 In a series of native kidney biopsies from patients with sarcoidosis, GIN was observed in 30–79%, while non-granulomatous AIN was observed in 14–44% of cases.9,10 Histologically, the granulomas of SLR are indistinguishable from those of systemic sarcoidosis. In our case, there was histological evidence of granulomatous SLR in the lymph node and in the bone marrow but not in the kidney. In a similar way that non-granulomatous AIN can be observed in systemic sarcoidosis, is not surprising to find AIN without granuloma, in systemic SLR, as described in our case.
In conclusion, the exceptional feature of our case is the significant impairment of kidney function observed in the post- ASCT period associated with a systemic granulomatous disease. After lymphoma relapse is ruled out, AIN secondary to SLR should be considered as a potential diagnosis. Permanent immunodeficiency secondary to chemotherapy exposure is frequently overlooked but can be a valuable clue to this rare condition. A kidney biopsy is crucial for an accurate diagnosis, which may prompt the initiation of corticosteroids to guarantee a sustained improvement of kidney function.
Footnotes
Acknowledgements
NA
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical approval (include full name of committee approving the research and if available mention reference number of that approval)
The study was approved by the Brazilian Ethics Committee (registration number 1978/21).
Informed consent
The patient signed an informed consent for these case report.
Trial registration (where applicable)
NA.
Guarantor
(The guarantor is the person willing to take full responsibility for the article, including for the accuracy and appropriateness of the reference list. This will often be the most senior member of the research group and is commonly also the author for correspondence. Please use initials only): V.T.C.S.
Contributorship
Report and review idea and study design: VTCS, LGL; data acquisition: VTCS, LGL, FLS, LBV, ICS; supervision or mentorship: VTCS, AMK, RSS, RAC, FZM, ECC. Each author contributed important intellectual content during manuscript drafting or revision and agrees to be personally accountable for the individual’s own contributions and to ensure that questions pertaining to the accuracy or integrity of any portion of the work, even one in which the author was not directly involved, are appropriately investigated, and resolved, including with documentation in the literature if appropriate.