
Abstract
Adenosine 5′-triphosphate acts as an extracellular signalling molecule (purinergic signalling), as well as an intracellular energy source. Adenosine 5′-triphosphate receptors have been cloned and characterised. P1 receptors are selective for adenosine, a breakdown product of adenosine 5′-triphosphate after degradation by ectonucleotidases. Four subtypes are recognised, A1, A2A, A2B and A3 receptors. P2 receptors are activated by purine and by pyrimidine nucleotides. P2X receptors are ligand-gated ion channel receptors (seven subunits (P2X1-7)), which form trimers as both homomultimers and heteromultimers. P2Y receptors are G protein-coupled receptors (eight subtypes (P2Y1/2/4/6/11/12/13/14)). There is both purinergic short-term signalling and long-term (trophic) signalling. The cloning of P2X-like receptors in primitive invertebrates suggests that adenosine 5′-triphosphate is an early evolutionary extracellular signalling molecule. Selective purinoceptor agonists and antagonists with therapeutic potential have been developed for a wide range of diseases, including thrombosis and stroke, dry eye, atherosclerosis, kidney failure, osteoporosis, bladder incontinence, colitis, neurodegenerative diseases and cancer.
Introduction
The potent actions of adenine compounds were first described by Drury and Szent-Györgyi in 1929. Years later, adenosine 5′-triphosphate (ATP) was proposed as the transmitter responsible for non-adrenergic, non-cholinergic transmission in the gut and bladder, and the term ‘purinergic’ was introduced by Burnstock in 1972. Early resistance to this concept was understandable since ATP was recognised first for its intracellular roles in many biochemical processes and the intuitive feeling was that such a ubiquitous and simple compound was unlikely to be utilised as an extracellular messenger. However, enzymes involved in the breakdown of extracellular ATP had already been described by the mid-1990s (see Yegutkin, 2008).
The concept of purinergic neurotransmission and the potent actions of extracellular ATP on many different cell types implied that there were purinergic membrane receptors. Purinergic receptors were described first in 1976 (Burnstock, 1976). Two years later, a basis for distinguishing two families of purinoceptors was proposed, identifying P1 and P2 receptors (for adenosine and ATP/adenosine 5′-diphosphate (ADP), respectively; Burnstock, 1978). Soon after, two subtypes of the P1 (adenosine) receptor were recognised (Londos et al., 1980; Van Calker et al., 1979). In 1985, a pharmacological basis for distinguishing two subtypes of P2 receptor (P2X and P2Y) was proposed (Burnstock and Kennedy, 1985). In the early 1990s, P1 (adenosine) receptors were cloned and characterised, and four subtypes were recognised (see Fredholm et al., 2001). In 1993, the first G protein-coupled P2Y receptors were cloned (Lustig et al., 1993; Webb et al., 1993) and a year later two P2X ligand-gated ion channel receptors (Brake et al., 1994; Valera et al., 1994). In 1994, it was proposed, on the basis of molecular structure and transduction mechanisms, that purinoceptors should belong to these two major families (Abbracchio and Burnstock, 1994). Currently, seven P2X receptor subtypes and eight P2Y receptor subtypes are recognised, including receptors that are sensitive to pyrimidines as well as purines (Abbracchio et al., 2006; North, 2002; Ralevic and Burnstock, 1998).
Purinergic signalling appears to be a primitive evolutionary system (see Verkhratsky and Burnstock, 2014). Many non-neuronal as well as neuronal mechanisms, including immune responses, exocrine and endocrine secretion, inflammation, pain, platelet aggregation and endothelial-mediated vasodilatation, involve purinergic signalling in mammals (Burnstock, 2006; Burnstock and Knight, 2004). Cell proliferation, differentiation and death that occur in development and regeneration are also mediated by purinergic receptors (Abbracchio and Burnstock, 1998; Burnstock and Verkhratsky, 2010). Reviews describe the history of the development of purinergic signalling and discuss future developments (Burnstock, 2012, 2014).
P1 receptors
Complementary DNAs encoding for two P1 receptor subtypes (A1 and A2) were isolated in 1989 (Libert et al., 1989). Soon after, the A3 subtype was identified (Zhou et al., 1992). Four different P1 receptor subtypes, A1, A2A, A2B and A3, were cloned and characterised in the early 1990s (see Fredholm et al., 2001). Polymorphisms have been observed in the A1 and the A2A receptors (Kumral et al., 2012). P1 receptors couple to adenylate cyclase; A1 and A3 are negatively coupled to adenylate cyclase, while A2A and A2B are positively coupled to adenylate cyclase (Reshkin et al., 2000). P1 subtype-selective agonists and antagonists have been identified (see Table 1). P1 receptor subtypes mediate diverse physiological effects, including modulation of cardiovascular, immune and central nervous system (CNS) activities (Ledent et al., 1997; Sun et al., 2001).
Characteristics of purine-mediated receptors.

Source: Updated from Burnstock (2003), with permission from Elsevier.
A3P5P: adenosine-3′-5′-bisphosphate; ADP: adenosine 5′-diphosphte; ADPβS: adenosine-5′-(β-thio)-diphosphate; 5′-AMPS: 5′-O-thiomnophosphate; Ap4A: diadenosine tetraphosphate; Ap5(γβ): adenosine pentaphosphate (βγ); ATPγS: adenosine-5′-(γ-thio)-triphosphate; ATP: adenosine 5′-triphosphte; ATPγS: adenosine-5′-(γ-thio)-triphosphate; BBG: brilliant blue green; 5-BDBD: 5-(3-bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-one; BzATP: 2′(3′)-O-(4-benzoylbenzoyl) adenosine 5′-triphosphate; β,γ-CF2ATP: α,β-difluoromethylene-ATP; cAMP: cyclic AMP; CCPA: chlorocyclopentyl adenosine; 2-Cl-IB-MECA: 2-chloro-N6-(3-iodobenzyl)-9-[5-(methylcarbamoyl)-β-
P2X receptor subtype agonist potencies are based on rat preparations, while P1 and P2Y receptor subtype agonist potencies are based on human preparations (=, equal potency; >, greater potency; ⩾, greater than or equal potency).
P2X receptors
P2X1-7 receptors show a subunit topology of intracellular N- and C- termini possessing consensus binding motifs for protein kinases and two transmembrane-spanning regions (TM1 and TM2). TM1 is involved with channel gating and TM2 lining the ion pore. There is a large extracellular loop, with 10 conserved cysteine residues (see Alves et al., 2014; North, 2002; Rokic and Stojilkovic, 2013). The stoichiometry of P2X1-7 receptors involves three subunits, which form a stretched trimer or a hexamer of conjoined trimers (Nicke et al., 1998; North, 2002; Stelmashenko et al., 2012). Heteromultimers as well as homomultimers are involved in forming the trimer ion pore (Nicke et al., 1998), including P2X2/3, P2X1/2, P2X1/5, P2X2/6, P2X4/6 and P2X1/4 receptors. Advances have been made by the use of knockout mice for P2X1, P2X2, P2X3, P2X4 and P2X7 receptors and transgenic mice that overexpress the P2X1 receptor. The association of various diseases with P2X receptor polymorphisms has been described (see Caseley et al., 2014).
P2X receptor subtypes
A complementary DNA (cDNA) encoding the P2X1 receptor was made from rat vas deferens (Valera et al., 1994). The agonist actions of ATP by α,β-methylene ATP (α,β-meATP) distinguish P2X1 and P2X3 receptors from the other homomeric forms. Several antagonists have been recognised to be selective for P2X1 receptors (see Table 1). Adenoviral expression of a P2X1 receptor-green fluorescent protein construct shows the receptor to be localised in clusters in vas deferens, with larger clusters apposing nerve varicosities (Burnstock, 2007).
Rat P2X2 receptor cDNA was isolated from PC12 cells (Brake et al., 1994) and human receptor cDNA from pituitary gland (Lynch et al., 1999). No agonists are currently known at present that are selective for P2X2 receptors. However, protons and low concentrations of zinc and copper potentiate P2X2 receptors. Antagonists for P2X2 receptors are shown in Table 1. The P2X2 receptor is non-desensitising, compared with the P2X1 and P2X3 receptors.
P2X1/2 heteromultimer receptors have been described in de-folliculated Xenopus oocytes (Brown et al., 2002; Calvert and Evans, 2004). pH sensitivity is characteristic of heteromeric P2X1/2 ion channels.
P2X3 receptor subunit cDNAs were taken from rat dorsal root ganglion cDNA libraries (Chen et al., 1995; Lewis et al., 1995), from a human heart cDNA library (Garcia-Guzman et al., 1997) and a zebrafish library (Egan et al., 2000). The antagonist NF023 is about 20 times less effective at P2X3 compared to P2X1 receptors (see Table 1). P2X3 receptors are prominently expressed on sensory neurons, including nociceptive nerve endings (Burnstock, 2003).
P2X2/3 heteromer receptors have been described (Lewis et al., 1995; Spelta et al., 2003). They exhibit a sustained current elicited by α,β-meATP. However, like homomeric P2X2 receptors, they are potentiated by low pH and, in common with the homomeric P2X3 receptor, they are very sensitive to block by 2′(3′)-O-(2,4,6-trinitrophenyl) ATP. P2X2/3 receptors are expressed by subpopulations of sensory neurons, sympathetic ganglion cells and brain neurons.
cDNAs for rat P2X4 receptors were isolated from both superior cervical ganglion (SCG) and brain (Bo et al., 1995; Buell et al., 1996). cDNAs for human, mouse, chick and Xenopus have also been isolated. P2X4 receptors are activated by ATP, but not by α,β-meATP. A useful distinguishing feature of ATP-evoked currents at P2X4 receptors is their potentiation by ivermectin. Unusual among the P2X receptors, the rat P2X4 receptor shows relative insensitivity to blockade by the conventional non-selective antagonists suramin and pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS). Carbamazepine derivatives have been claimed to be potent P2X4 receptor antagonists (Tian et al., 2014; see Table 1). Currents evoked by ATP at the mouse P2X4 receptor are increased by PPADS and suramin, probably because they are also ectonucleotidase inhibitors (Burnstock, 2003).
P2X1/4 heteromeric receptors were shown to have kinetic properties resembling homomeric P2X4 receptors and a pharmacological profile similar to homomeric P2X1 receptors (Nicke et al., 2005). An important advance was made when the crystal structure of the P2X4 receptor was described (Kawate et al., 2009; Figure 1).
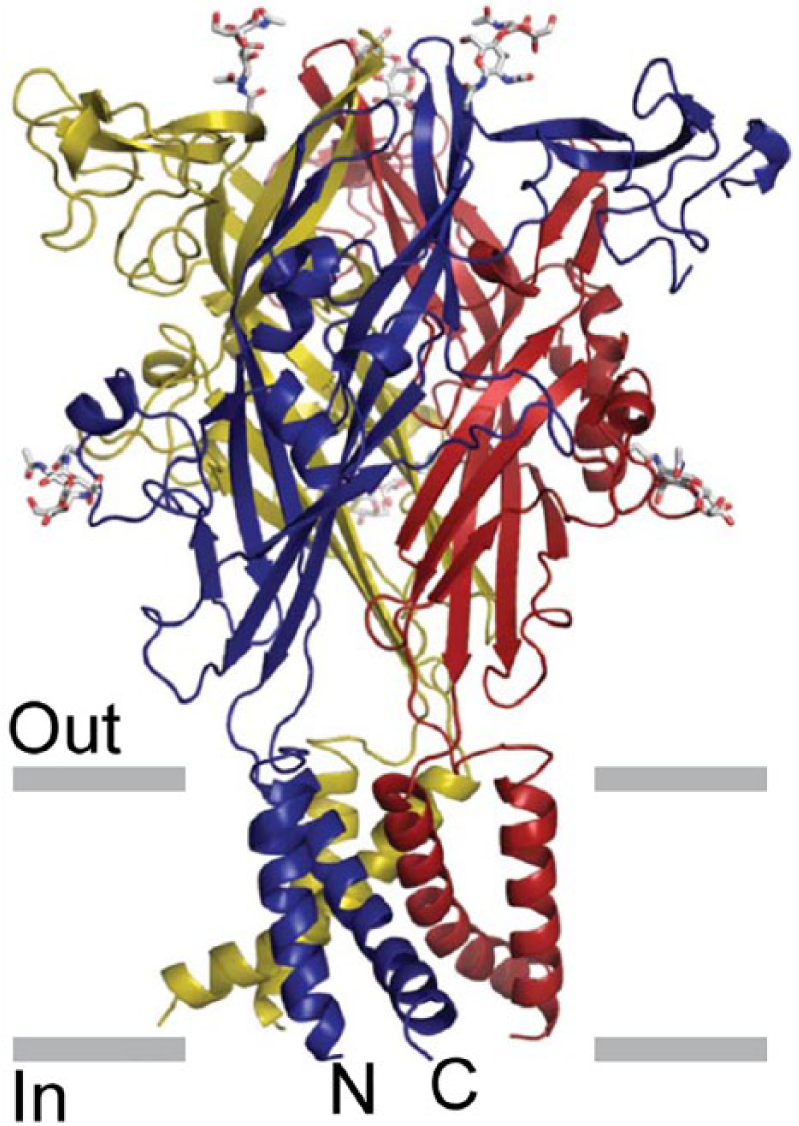
The architecture of the P2X4 receptor. Stereoview of the homotrimeric ΔzfP2X4 structure viewed parallel to the membrane. Each subunit is depicted in a different colour. N-acetylglucosamine (NAG) and glycosylated asparagine residues are shown in stick representation. The grey bars suggest the boundaries of the outer (out) and inner (in) leaflets of the membrane bilayer.
P2X5 receptor cDNA was first isolated from rat coeliac ganglion and heart (Burnstock, 2003; Garcia-Guzman et al., 1996). It was also cloned from embryonic chick skeletal muscle (Bo et al., 2000) and a bullfrog P2X5 receptor from larval skin. Human P2X5 receptor cDNAs are missing exon 10 (hP2X5a) or exons 3 and 10 (P2X5b) (Roberts et al., 2006; Stojilkovic et al., 2005). Currents elicited by ATP in cells expressing the rat P2X5 receptor are of small amplitude, compared with the currents observed with P2X1, P2X2, P2X3 or P2X4 receptors. P2X5 mRNA is highly expressed in developing skeletal muscle (Burnstock, 2003).
The P2X1/5 heteromer is characterised by a sustained current evoked by α,β-meATP, which does not occur for either of the homomers when expressed separately (Burnstock, 2007; Surprenant et al., 2000). Cells expressing P2X1/5 receptors are more sensitive to ATP than those with homomeric receptors. P2X2/5 heteromeric receptors have also been recognised (Compan et al., 2012).
The rat P2X6 receptor was cloned from SCG (Burnstock, 2007) and rat brain (Soto et al., 1996). The human equivalent was isolated from peripheral lymphocytes and was abundantly expressed in human and mouse skeletal muscle. The P2X6 subunit is only functionally expressed as a heteromultimer.
Heteromeric P2X2/6 receptors were expressed in HEK293 cells (Torres et al., 1999). The most convincing difference in oocytes expressing P2X2/6 receptors compared to those expressing only P2X2 receptors is that at pH 6.5 the inhibition of the current by suramin is biphasic (King et al., 2000). P2X2/6 receptors are expressed particularly by respiratory neurons in the brain stem.
P2X4/6 heteromeric receptors were described in oocytes (Khakh et al., 1999). The P2X4/6 heteromer differs only in minor respects from that of P2X4 homomers. They are prominently expressed in adult trigeminal mesencephalic nucleus and in hippocampal CA1 neurons (North, 2002).
A chimeric cDNA encoding the rat P2X7 receptor was first isolated from SCG and medial habenula. Full-length cDNAs were later isolated from a rat brain cDNA library (Surprenant et al., 1996). The unique feature of the P2X7 receptor is that in addition to the usual rapid opening of the cation-selective ion channel, with prolonged exposure to high concentrations of ATP it undergoes a channel to pore conversion to allow the passage of large dye molecules such as ethidium and YO-PRO-1. This often leads to apoptotic cell death. 2′,3′-O-(benzoyl-4-benzoyl)-ATP (BzATP) is a potent, although not selective, agonist at the P2X7 receptor. After continuous application of BzATP (30 µM) for about 30 s, the plasma membrane develops large blebs. Blebs are usually preceded by the shedding of smaller vesicles (<1 µm diameter) that release inflammatory cytokines. A number of potent antagonists have been developed (see Table 1).
P2Y receptors
P2Y1 and P2Y2 receptors were cloned in 1993 (Lustig et al., 1993; Webb et al., 1993). Since then several other subtypes have been isolated by homology cloning. The Gi-coupled ADP receptor (P2Y12) of platelets was isolated by expression cloning in 2001 (Hollopeter et al., 2001). P2Y13 and P2Y14 receptors were characterised during a study of orphan receptors (Chambers et al., 2000; Communi et al., 2001a). At present, there are eight recognised human P2Y receptors: P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13 and P2Y14 (Abbracchio et al., 2003, 2006; see Table 1). The missing numbers represent either non-mammalian orthologs or receptors which have no functional evidence of responsiveness to nucleotides. A p2y8 receptor was cloned from the frog embryo, which was shown to be involved in the development of the neural plate (Bogdanov et al., 1997). A P2Y-like receptor, GPR17, has been described (Parravicini et al., 2008). Two distinct P2Y receptor subgroups characterised by a relatively high level of sequence divergence have been identified (Abbracchio et al., 2006). The first subgroup includes P2Y1,2,4,6,11 and the second subgroup includes the P2Y12,13,14 subtypes (see Figure 2). For some of the P2Y receptor subtypes, potent and selective synthetic agonists and antagonists have been identified (see Table 1). Activation of several P2Y receptors is associated with the stimulation of mitogen-activated protein kinase (MAPK; Abbracchio et al., 2006). Ion channel couplings of P2Y receptors are primarily of importance in neurons, but they have been detected in various other tissues, including cardiac muscle cells (Abbracchio et al., 2006; Vassort, 2001). Polymorphisms of P2Y1, P2Y2 and P2Y12 receptors have been identified associated with various diseases (Läer et al., 2013; Lev et al., 2007; Wesselius et al., 2013). There is dimerisation involving P2Y receptors with non-P2Y receptors, for example, rat P2Y1 and adenosine A1 receptors in neurons, in rat cortex, hippocampus and cerebellum (Yoshioka et al., 2002). Functional P2Y1 and P2Y2 receptors, co-localised at the neuromuscular junctions with nicotinic acetylcholine (ACh) receptors, have been reported in mammalian, chicken and amphibian muscles.

(a) Dendrogram to show relatedness of 29 P2X receptor subunits. Full-length amino acid sequences were aligned with Clustal W using default parameters. The dendrogram was constructed with TreeView. h, human (Homo sapiens); r, rat (Rattus norvegicus); m, mouse (Mus musculus); gp, guinea pig (Cavia porcellus); c, chicken (Gallus gallus); zf, zebrafish (Danio rerio); bf, bullfrog (Rana catesbeiana); x, claw-toed frog (Xenopus laevis); f, fugu (Takifugu rubripes). The ellipses indicate the apparent clustering by relatedness into subfamilies. Source: Reproduced from North (2002), with permission from the American Physiological Society. (b) A phylogenetic tree (dendrogram) showing the relationships among the current members of the P2Y receptor family (human P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12 and P2Y13 receptors) and the human UDP-glucose receptor (here indicated as the P2Y14 receptor). The P2Y receptors can be divided into two subgroups shown with green and lilac backgrounds. Sequences were aligned using CLUSTALX and the tree was built using the TREEVIEW software. Source: Reproduced from Abbracchio et al. (2003), with permission from Elsevier.
P2Y receptor subtypes
P2Y1 receptors have been cloned and characterised in human, rat, mouse, cow, chick, turkey and Xenopus tissues. ADP is a more potent agonist than ATP and 2-methylthio ADP is even more potent. The most effective antagonists to display selectivity for the P2Y1 receptor at present are MRS2179, MRS2279 and MRS2500 (see Table 1). P2Y1 mRNA is high in different regions of the human brain, prostate gland and placenta (Burnstock and Knight, 2004; Léon et al., 1996). In post-mortem brain sections from Alzheimer’s disease patients, the P2Y1-like immunoreactivity in the hippocampus and entorhinal cortex was localised to neurofibrillary tangles, neuritic plaques and neuropil threads (Moore et al., 2000).
P2Y2 receptors have been cloned and characterised from human, rat, mouse, canine and porcine cells or tissues (Shen et al., 2004). P2Y2 receptors are activated by equivalent concentrations of ATP and UTP. The γ-thiophosphate, UTPγS and INS 37217 are potent hydrolysis-resistant agonists of P2Y2 receptors (Abbracchio et al., 2006). Current P2Y2 receptor antagonists are shown in Table 1. Expression of P2Y2 receptor mRNA has been described in many tissues (Burnstock and Knight, 2004). P2Y2 receptor expression in smooth muscle cells is up-regulated by inflammatory agents, including interleukin-1β, interferon-γ and tumour necrosis factor-α (Hou et al., 2000). In epithelial cells, P2Y2 receptor activation increases Cl− secretion and inhibits Na+ absorption (Kellerman et al., 2002). A P2Y2 receptor knockout mouse has been described that is defective in nucleotide-stimulated ion secretion in airway epithelial cells (Cressman et al., 1999). P2Y2 receptors inhibit bone formation by osteoblasts (Hoebertz et al., 2002) and N-type calcium currents in neurons (Brown et al., 2000).
P2Y4 receptors have been cloned and characterised from human, rat and mouse. UTP is the most potent activator of the human P2Y4 receptor (Communi et al., 2004). However, the recombinant rat and mouse P2Y4 receptors are activated equipotently by ATP and UTP (Bogdanov et al., 1998). Reactive Blue 2 blocks rat P2Y4 receptors, but only partially blocks human P2Y4 receptors. P2Y4 mRNA and protein was most abundant in the intestine of both mouse and humans, but has also been described in other organs (Burnstock and Knight, 2004). P2Y4-null mice show normal behaviour, growth and reproduction; however, the chloride secretory response to apical UTP and ATP of the jejunal epithelium was abolished (Robaye et al., 2003).
P2Y6 receptors are selective for UDP in mouse, rat and human (Lazarowski et al., 2001). Selective agonists and antagonists are described in Table 1. A wide tissue distribution of P2Y6 mRNA and protein has been reported, and the highest expression was in spleen, intestine, liver, brain and pituitary (Burnstock and Knight, 2004).
The human P2Y11 receptor has a unique profile (Abbracchio et al., 2006). The hP2Y11 receptor gene differs from other P2Y receptor subtype genes having the presence of a 1.9-kb intron in the coding sequence that separates an exon encoding the first 6 amino acid residues from a second exon encoding the remaining part of the protein (Communi et al., 2001b).
P2Y12 receptors have been identified and characterised in human, rat and mouse (Abbracchio et al., 2006). ADP is the agonist of this receptor. The P2Y12 receptor is highly expressed in platelets where it is the molecular target of antiplatelet drugs, clopidogrel and ticagrelor (Savi and Herbert, 2005). The P2Y12 receptor is also expressed in sub-regions of the brain, glial cells, brain capillary endothelial cells, smooth muscle cells and chromaffin cells (Burnstock and Knight, 2004). P2Y13 receptors have been identified and characterised in human, mouse and rat (Abbracchio et al., 2006). Naturally occurring agonists of the P2Y13 receptor are ADP and di-adenosine triphosphate. Selective antagonists of the human P2Y13 receptor are shown in Table 1. The P2Y13 receptor is expressed in spleen, placenta, liver, heart, bone marrow, monocytes, T cells, lung and various regions of the brain (Fumagalli et al., 2004).
The P2Y14 receptor is 47% identical to the P2Y12 and P2Y13 receptors. The gene for this receptor is in human chromosome 3q24-3q25 (Abbracchio et al., 2003). The P2Y14 receptor is activated by UDP-glucose and is coupled to the Gi/o family of G proteins (Harden, 2004). P2Y14 mRNA is distributed in many systems in the human body. Chemoattractant and neuroimmune functions have been claimed for the P2Y14 receptor.
Physiology of purinergic signalling
Early studies were focused largely on short-term purinergic signalling in neurotransmission, neuromodulation, secretion, chemoattraction and acute inflammation, but there is increasing interest in long-term (trophic) signalling involving cell proliferation, differentiation, motility and death in development, regeneration, wound healing, restenosis, epithelial cell turnover, cancer and ageing (Abbracchio and Burnstock, 1998; Burnstock and Verkhratsky, 2010). In blood vessels, there is dual purinergic short-term control of vascular tone by ATP (Burnstock, 2002): ATP released as a cotransmitter from perivascular sympathetic nerves activate P2X receptors resulting in contraction of smooth muscle; ATP released from endothelial cells during changes in blood flow (shear stress) and hypoxia acts on P2X and P2Y receptors on endothelial cells, resulting in production of nitric oxide and relaxation. There is also long-term control of cell proliferation and differentiation, migration and death involved neovascularisation, restenosis following angioplasty and atherosclerosis (Erlinge and Burnstock, 2008). Purinergic signalling is involved in development, ageing and regeneration (Burnstock, 2007). Purinergic receptors are expressed on stem cells (Burnstock and Ulrich, 2011; Delic and Zimmermann, 2010; Trujillo et al., 2009). Many cell types release ATP physiologically in response to gentle mechanical distortion, hypoxia or some agents (Bodin and Burnstock, 2001). The mechanism of ATP transport includes, in addition to vesicular release, ABC transporters, connexin or pannexin hemi-channels, maxi-ion channels and even P2X7 receptors (Burnstock, 2007; Lazarowski et al., 2011). Extracellular breakdown of released ATP is by ectonucleotidases, including E-NTPDases, E-NPPS, alkaline phosphatase and ecto-5′-nucleotidose (Yegutkin, 2008; Zimmermann et al., 2007). P1 and P2 receptors are involved in neurotransmission and neuromodulation in the CNS, and in normal behaviour, including memory, feeding, locomotion and cognition (Burnstock et al., 2011).
Purinergic pathophysiology and therapeutic potential
ATP was shown early to be a major cotransmitter with ACh in parasympathetic nerves mediating contraction of the urinary bladder of rodents (Burnstock et al., 1978). However, in healthy human bladder, the role of ATP as a cotransmitter is minor, but under pathological conditions, such as interstitial cystitis, outflow obstruction and most types of neurogenic bladder, the purinergic component is increased to about 40% (Burnstock, 2001, 2006). There is also a significantly greater cotransmitter role for ATP in sympathetic nerves in spontaneously hypertensive rats (Vidal et al., 1986). P2X3 receptors are located on small nociceptive sensory nerves and are involved in the initiation of pain (Bradbury et al., 1998; Burnstock, 1996, 2013; Chen et al., 1995). There are peripheral extensions of the sensory nerves in skin, tongue and visceral organs and central projections to inner lamina 2 of the spinal cord. A hypothesis describing purinergic mechanosensory transduction and pain in visceral organs was published in 1999 (Burnstock, 1999). It was proposed that ATP, released from urothelial/epithelial cells during distension, acts on P2X3 and P2X2/3 receptors on subepithelial sensory nerve endings to send nociceptive messages via sensory ganglia to the pain centres in the brain (Burnstock, 1999). Supporting evidence has been reported in the bladder (Vlaskovska et al., 2001), ureter (Rong and Burnstock, 2004) and gut (Wynn and Burnstock, 2006). Purinergic mechanosensory transduction via low threshold fibres is also involved in urine voiding (Cockayne et al., 2000). Antagonist to P2X4, P2X7 and P2Y12 receptors on microglia have been shown to reduce neuropathic and inflammatory pain (Burnstock, 2009; Inoue, 2007). There is increasing attention to the potential roles of purinergic signalling in trauma, ischaemia and neurodegenerative conditions, including Alzheimer’s, Parkinson’s and Huntington’s diseases, multiple sclerosis and amyotrophic lateral sclerosis (Burnstock, 2007; Sperlágh and Illes, 2014). The involvement of purinergic signalling in epilepsy, neuropsychiatric diseases and mood disorders has also been reported (Boisson and Burnstock, 2010; Burnstock, 2008b).
Purinergic agents are being investigated for the treatment of disorders of the urinary tract (Burnstock, 2011), skeletal muscle (Ryten et al., 2004), gut (Burnstock, 2008a), bone (Burnstock and Arnett, 2006; Orriss et al., 2010), the cardiovascular system (Erlinge and Burnstock, 2008), kidney (Bailey et al., 2007; Taylor et al., 2009) and the reproductive system (Calvert et al., 2008; Gür et al., 2009). There are also reports that extracellular ATP acts on P2Y2 receptors to facilitate HIV-1 infection (e.g. Séror et al., 2011). The therapeutic potential of purinergic compounds for the treatment of cancer is being explored (Burnstock and Di Virgilio, 2013; Shabbir and Burnstock, 2009; Shabbir et al., 2008; Stagg and Smyth, 2010; White and Burnstock, 2006; White et al., 2009). Selective purinoceptor agonists and antagonists with therapeutic potential are also being developed for thrombosis and stroke, atherosclerosis, kidney failure, osteoporosis, bladder incontinence and colitis (Bartlett et al., 2014; Burnstock, 2006, 2013). This would be facilitated by the discovery of selective purinoceptor agonists and antagonists that are orally bioavailable and stable in vivo.
Footnotes
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author received no financial support for the research, authorship and/or publication of this article.