
Abstract
Research investigating epigenetics and chromatin function in brain and behaviour has mushroomed over the last two decades. And yet epigenetics as a biological concept predates the discovery in the 1950s of DNA as the principle mode of inheritance by over a decade. This review explores the past, present and future research into epigenetics as it relates to understanding brain development and function
Introduction
Research investigating epigenetics and chromatin function in brain development and function has mushroomed over the last two decades. And yet epigenetics as a biological concept predates the discovery in the 1950s of DNA as the principle mode of inheritance by over a decade.
At the most fundamental level, epigenetic control is exerted through the covalent modification cytosine residues leading to 5-methylcytosine (5mC). This process, known as DNA methylation (DNA-me), is central to transcriptional regulation, transposable element suppression, genomic imprinting, X-chromosome inactivation and the stability of the genome (Shin et al., 2014). In mammals, the majority of DNA-me is found at CpG dinucleotides as part of what are termed CpG islands. These CpG islands often form the differentially methylated regions (DMRs), which provide the key, often stable, epigenetic marks across the life cycle of mammals. Nonetheless, there is now an increasing awareness of the importance of non-CpG DNA-me at so-called CpH sites (H = A, C or T) and also of hydroxymethylation (5hmC) for brain function (Guo et al., 2014; Kriaucionis and Heintz, 2009).
In addition to direct modification of the DNA, gene regulation can also be exerted via modifications to the three-dimensional (3D) structure and packaging of DNA and its associated histone proteins and factors (Qureshi and Mehler, 2014). The DNA within a cell is far from naked; first it is wrapped around a histone octamer to form a nucleosome, which is in turn connected together via DNA wrapped around linker histones such as H1. These are then packed and arrayed into progressively higher-order chromatin structures. Generally, chromatin states are divided into euchromatin (open) and heterochromatin (closed). However, this may be over-simplistic as chromatin structure is very dynamic with the accessibility being determined by modifications (acetylation, methylation, phosphorylation, to name but a few) of individual histone proteins, nucleosome movement and over larger genomic regions.
Here, I provide an overview of history of epigenetics research in relation to the brain and behaviour. As well as looking at the past- and present-day research into epigenetics, I also touch upon possible future development of relevance to neuroscience in this field.
Past
The idea of ‘epigenetics’ was first championed in the 1940s by Conrad Waddington (1942), an embryologist interested in the mechanisms of cell differentiation and subsequent maintenance of cellular identity. He correctly reasoned that as all cells carry the same genetic information there must be another layer of information over and above the genetic information which allows a totipotent cell to become specialized and to stay specialized with subsequent cell divisions. Put more generally, epigenetics refers to the process modulating the expression of a genotype into a given phenotype (Waddington, 1942). This classical definition of epigenetic mechanisms is of course completely applicable to the development of the brain and, by default, the development of behaviour.
Perhaps because of its consequences for the expression of phenotypes, one of the first aspects of epigenetics directly linked with the brain and behaviour was genomic imprinting. Genomic imprinting is the process by which the inherited parental genomes are epigenetically marked differentially in the germline, initially by DNA-me but built upon by modification of associated chromatin modification (Figure 1). This results in expression of some imprinted genes only from the maternal copy (allele), whereas others are solely expressed from the paternal allele. Parental-specific monoallelic expression of this kind is highly unusual (most genes are biallelic, being expressed from both inherited copies) and genomic imprinting is limited to mammals and only affects around 150 genes. However, correct expression of these genes is absolutely critical for normal development to progress, as evidenced by the death before mid-gestation of mouse embryos composed of solely maternally (parthenogenetic) or paternally (androgenetic/gynogenetic) derived genomes (Barton et al., 1984; McGrath and Solter, 1984). Soon after the discovery of the non-equivalence of maternal and paternal genomes, the first imprinted genes were identified and two distinct neurodevelopmental disorders were linked to a cluster of imprinted genes on human chromosome 15q11–q13 (Figure 1). Large deletions at this locus give rise to Angelman syndrome when maternally derived, but Prader–Willi syndrome when paternally derived (Williams et al., 1990). These clinical findings, coupled with the pioneering mouse work by Barry Keverne and colleagues in the 1990s (Keverne, 1997), set the scene for future studies into how changes in the expression and epigenetic regulation of this small subset of mammalian genes has consequences for a range of the brain and behavioural phenotypes.
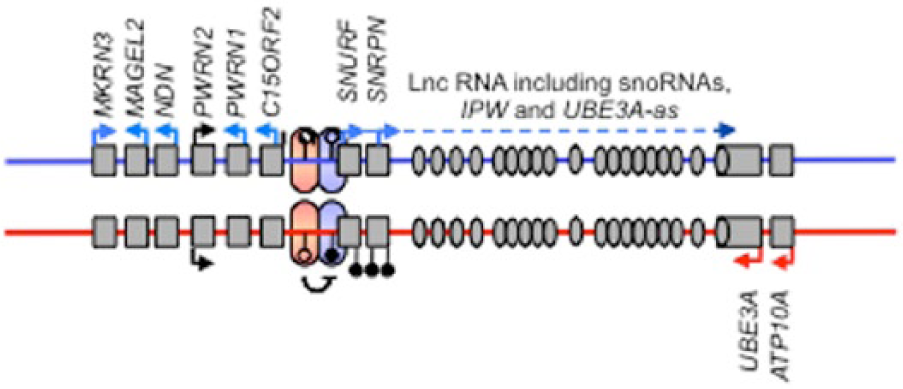
Schematic showing a representative imprinted gene cluster, in this case, the Angelman and Prader–Willi Syndrome (AS/PWS) imprinted cluster on human chromosome 15. As is common for imprinted genes, within this cluster are both maternally (red arrows) and paternally (blue arrows) expressed genes, including a long non-coding RNA (Lnc-RNA). Also marked with black ‘lollipops’ is the differentially methylated region (DMR), which in the case of the AS/PWS locus is methylated on the maternally derived chromosome and un-methylated on the paternal chromosome. The parental-specific marking of the DMR distinguishes the maternal and paternal chromosomes at this interval and guides parental-specific gene expression control via chromatin changes and non-coding RNA (e.g. UBE3A antisense, UBE3A-as).
As we shall see in the next section, in addition to the programmed developmental role, a more recent finding is that epigenetic mechanisms can also be labile and may act as a biochemical record of the impact of environmental events. However, this idea also has its origins some time ago. As far back as the late 1960s, the idea that DNA-me may provide a molecular mechanism underpinning memory formation was first postulated (Griffith and Mahler, 1969) and again highlighted as a possibility by Francis Crick (1984). Much more recent work by David Sweatt and colleagues has finally confirmed this inkling, showing that DNA-me associated with the memory-suppressor gene calcineurin in the medial prefrontal cortex persists for at least 30 days following fear conditioning (Miller et al., 2010), suggesting, in this brain region at least, that this epigenetic change is an additional layer of molecular information and a marker for memory.
Present
Despite its long history and the prescient insight of a number of pioneering biologists, the huge progress in understanding the role of epigenetic mechanisms in the brain accelerated over last 20 years, driven by the development of tools to study the various biochemical processes involved. There are increasingly sophisticated molecular and biochemistry techniques for quantifying and modifying the different, but interacting, epigenetic mechanisms including DNA-me, histone modifications and varied classes of regulatory non-coding RNAs.
Neuroscientists have applied these tools to understanding the processes involved in neurodevelopment and brain maturation. A good example is the epigenetic mechanisms underlying the development of the brain and behaviour phenotypes exhibited by different castes of social insects. Social insects are characterized by a division of reproductive labour whereby some group members are non-reproductive workers who provision and raise the offspring of their queen, whose sole function is reproduction (Wilson, 1971). These castes exhibit highly individual morphological and behavioural phenotypes, despite often sharing >75% of their genes. The differentiation of larvae into different castes has been compared to the cellular lineage reprogramming of mammals (Patalano et al., 2012), and a number of studies have demonstrated differences in global brain levels of DNA-me between distinct castes of social insects including in bees and ants (Yan et al., 2014). Critically, the causal importance of this epigenetic mark has been confirmed by showing that manipulating the levels of a key DNA-me enzyme in honeybee larvae leads to an increase in the number of queens produced (Kucharski et al., 2008).
In mammals much of the research into the epigenetic control of neurodevelopment has taken a more reductionist approach, focusing on the key changes that underpin the process of specialization of pluripotent cells into neurons (Hirabayashi and Gotoh, 2010). Predictably, the number of genes that require regulation during neurogenesis is large and involves the coordination of a great many regulatory processes. The level of DNA-me is certainly important, as indicated by changes in DNA-me associated with the imprinted gene Dlk1, resulting in a switch from paternal-only monoallelic, to biallelic expression during postnatal neurogenesis (Ferron et al., 2011). However, at present the key general mechanisms for cell fate during neurogenesis appear to involve changes in chromatin structure and transcriptional valiance, via histone modifications, polycomb groups and other chromatin modifiers (Tyssowski et al., 2014).
Interestingly, a great deal of our understanding of the importance of such epigenetic mechanisms for human neurodevelopment has come about through genomic studies. Perhaps unsurprisingly, we now recognize that several rare neurodevelopmental disorders involve mutations in genes encoding proteins involved in setting, interacting with or reading the normal epigenetic marks spread throughout the genome during development. Mutations in these single genes can result in disrupted and incorrect expression of hundreds of other genes during the course of development. Selected examples include Rett, Kleefstra, Rubinstein–Taybi, Angelman and Prader–Willi syndromes (Isles, 2015). More recently the first specific mutations causing schizophrenia were identified in the gene SETD1A, which encodes a histone methyltransferase (Singh et al., 2016).
In addition to these very rare and highly penetrant mutations, detailed analyses of the common but low penetrance genetic variants, as identified by genome-wide association studies (GWASs), have also implicated abnormalities in epigenetic processes in psychopathology. For instance, by assigning the GWAS calls associated with schizophrenia to biological pathways, bioinformaticians are able to detect which particular biological processes are significantly enriched within the signal. Such studies provide compelling evidence that genes involved in histone modification and/or chromatin remodelling are implicated in the development of psychiatric illness (McCarthy et al., 2014; Network Pathway Analysis Subgroup of Psychiatric Genomics, 2015; Singh et al., 2016).
Over the last decade or so, a more controversial area of behavioural and neural epigenetics has emerged whereby the progress in molecular biology techniques has been used to identify changes in DNA-me and/or chromatin in response to environmental effects. There are many robust epidemiological studies linking environmental insults with the development of neuropsychiatric illness and/or changes in behaviour, such as prenatal exposure to famine and schizophrenia incidence (Susser and Lin, 1992). Changes to the epigenome could be a mechanism by which some of these environmental effects are mediated (Isles and Wilkinson, 2008). As a consequence, a number of groups have begun to measure genome-wide levels of DNA-me and ask whether epigenetic differences can disentangle the relationship between disease states and/or exposure to environmental insults, such as prenatal diet and attention deficit disorder (Rijlaarsdam et al., 2017). However, it is currently impossible to determine the causal relationship in these studies; are any observed changes in DNA-me simply an epigenetic reaction to the disorder? Furthermore, while some of the observed epigenetic changes are significant statistically, they are quantitatively small, raising questions of their biological relevance. Finally, by necessity these human studies often profile the epigenome of blood samples, raising the question of the relevance of any potential changes to brain function (Hannon et al., 2015).
More progress has been made in this area through the use of animal and/or cellular models, where there is access to appropriate tissues and experimental manipulations allow the true causal relationships to be determined. Here, studies have demonstrated the causative epigenetic links between heat exposure and thermotolerance (Kisliouk and Meiri, 2009), drug use and addiction (Kumar et al., 2005), and the level of maternal care and future stress reactivity (Weaver et al., 2004).
Future
One of the key challenges facing many aspects of neuroscience, and epigenetics research in particular, is the heterogeneity of cell types in the brain. Unlike genetic studies where, generally, the same DNA can be found samples from any tissue, epigenetic changes may be localized to specific cells. This has obvious implications for the relevance of human epigenome–wide association studies (EWASs) where primary tissue may not be available (Hannon et al., 2015). But these issues are also pertinent for in vivo animal studies of epigenetics in the brain. However, as sequencing techniques become increasingly sophisticated and cost-effective, the idea of single-cell epigenomic studies is on the horizon (Clark et al., 2016). For instance, coupled with single-unit electrophysiological recordings, such techniques may allow fully integrated epigenome and transcriptome profiling (Figure 2) of synaptic plasticity.

Schematic illustrating the possible future applications of single-cell epigenomics to neuroscience. The example here links single-cell electrophysiological recordings with single-cell transcriptomics, genomics and epigenomics. For instance, this would enable a true readout of the gene expression and associated DNA-me and/or chromatin changes that underpin synaptic plasticity.
In addition to ever-finessed measurements of changes in the epigenome and transcriptome, there will also be opportunities in the future to manipulate the epigenome locally. This may well be achieved via optogenetic or genome editing techniques such as CRISPR-cas9 (Vojta et al., 2016) or a combination of both. Certainly, optogenetics has already been used to locally inhibit the REST neural gene transcription factor (Paonessa et al., 2016), and very recently expression of ΔFosB has been controlled in specific cell types within the nucleus accumbens using the histone methyltransferase G9a coupled to zinc finger proteins (ZFP; Hamilton et al., 2018). Clearly, the field of epigenome editing is already developing rapidly.
Conclusion
There is a great deal of interest in the role epigenetics may play in brain development and function. Like all research in the field of epigenetics, the excitement in relation to neuroscience has been largely driven by the rapid and increased availability of biochemical tools to interrogate changes in gene expression, DNA-me and chromatin modification. This has led to some fascinating findings, particularly in relation to understanding the development of the nervous system and how this translates into brain function.
Footnotes
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
A.R.I. is a member of the MRC Centre for Neuropsychiatric Genetics and Genomics (UK Medical Research Council, MR/L010305/1).