
Abstract
In 2000, with the completion of the human genome project, nine related channels were found to comprise the complete voltage-gated sodium gene family and they were renamed NaV1.1–NaV1.9. This millennial event reflected the extraordinary impact of molecular genetics on our understanding of electrical signalling in the nervous system. In this review, studies of animal electricity from the time of Galvani to the present day are described. The seminal experiments and models of Hodgkin and Huxley coupled with the discovery of the structure of DNA, the genetic code and the application of molecular genetics have resulted in an appreciation of the extraordinary diversity of sodium channels and their surprisingly broad repertoire of functions. In the present era, unsuspected roles for sodium channels in a huge range of pathologies have become apparent.
Introduction
Voltage-gated sodium channels (VGSCs) are essential components of electrical signalling in excitable tissues and have been linked to a vast and increasing range of disorders (Eijkelkamp et al., 2012). Their distinct biophysical properties and patterns of expression contribute to the functional repertoire of neurons and information processing that determine brain function. The combination of technical advances in genetics and electrophysiology resulted in the cloning of the first sodium channels in 1984, and since then our insights into the structure and function of sodium channels have increased dramatically. Here we look back to the era of animal electricity and forward to new aspects of sodium channel biology and exciting therapeutic prospects for diseases ranging from cancer to epilepsy, pain and autism.
Early studies of animal electricity
Electromagnetism and the use of the compass were known in antiquity, but the remarkable intellectual and experimental feats of the 18th century underpin our present understanding of electrical signalling in the nervous system. An interesting historical overview is provided by Piccolino (1997). Generation of electric currents by friction and charge storage in Leyden jars were already established when Galvani famously discovered in 1780 that frogs legs could be made to twitch when charged metals were applied to them. He described the existence of animal electricity that Volta later showed was not a reflection of a unique vital force, but could be imitated by currents produced from the first battery, the Voltaic pile. Volta subsequently received the Copley model from The Royal society in 1800 as well as many accolades across Europe. In London, these experiments were extended by a cousin of Galvani, Aldini, to electrically stimulate the corpse of an unfortunate criminal that twitched and opened an eye as a result. This gruesome observation led to much debate and eventually in 1816, to the remarkable novel ‘Frankenstein’ by Mary Shelly that has inspired the horror movie business for the last century.
After the discovery that there was a membrane potential across nerves, du Bois-Reymond described the action potential in 1848. Julius Bernstein was the first to expound a membrane theory that involved altered ion permeability to explain electrical propagation. In the mid-20th century, Kenneth Coles at Columbia showed that there was an increase in conductance associated with the action potential, developing the voltage clamp technique subsequently used to great effect by Hodgkin and Huxley (1945) to examine the role of different ion fluxes that contribute to action potential propagation. Thus, the stage was set by the mid-1950s for an electrophysiological analysis of electrical signalling in the nervous system, just as genetics matured into a molecular science with the discovery of the structure of DNA.
Purification and cloning of sodium channels
Toxins proved useful in both purifying sodium channels and in understanding the structural and molecular determinants of sodium channel gating (Hartshorne and Catterall, 1981). The bacterial toxins tetrodotoxin (TTX) and saxitoxin isolated from puffer fish and shell fish were found to exert their toxic action through sodium channels as shown by Narahashi in 1964. As saxitoxin could be tritiated, this allowed Catterall to purify binding proteins and gain insights into primary amino acid sequences associated with the channels. Dramatic developments in cloning followed the elucidation of the genetic code and the isolation of reverse transcriptase by both Temin and Baltimore that enabled cDNA to be copied from mRNA transcripts. With the purification of restriction endonucleases that allow DNA to be cut and pasted into bacteriophage vectors, the stage was set for the explosive development of molecular biology. This stunning set of technical advances allowed Noda and Numa first to show that multiple transcripts of sodium channels existed in the rat brain, second to isolate whole cDNA transcripts of mRNA and finally to express functional channels in xenopus oocytes (Noda et al., 1986). These technical feats have given us a wealth of insights into sodium channel function, particularly when coupled with the ability to genetically modified mice to through homologous recombination in embryonic stem cells, a technology that in the 21st century is being facilitated by the use of CRISPR-Cas 9 technology. The regulation of sodium channel transcription was also studied in the 1990s and groups led by Anderson and Mandel both identified a regulatory DNA sequence that plays an important role in restricting most sodium channel gene expression to neurons – the neuron-restricted silencing element (Chong et al., 1995; Schoenherr and Anderson, 1995).
Structure and activity
The VGSC gene family comprises nine homologous members SCN1A–SCN11A, while the encoded sodium selective ion channels are numbered from NaV1.1 to NaV1.9. Nax encoded by SCN6/7A, though structurally related to VGSCs, is activated by altered sodium concentrations and is physically associated with the sodium potassium ATPase (Noda and Hiyama, 2015). See Table 1.
Voltage-gated sodium channel subunits.

TTX: tetrodotoxin; CNS: central nervous system; PNS: peripheral nervous system.
Each α-subunit (~260 kDa) contains four homologous domains comprising six transmembrane segments (see Figure 1). One α-subunit is sufficient to form a functional channel but α-subunits associate with β-subunits (SCN1B–SCN4B), which modulate channel biophysics and trafficking. The voltage sensors contain repeated motifs of positively charged amino acids followed by hydrophobic residues arranged in an α-helix with a linear array of positively charged residues. Depolarisation of the cell alters the electric field across the cell membrane resulting in the rapid movement of the DI–III S4 voltage sensors and a conformational change in the protein which opens the ion channel pore. Channel opening caused by membrane depolarisation results in a rapid influx of sodium ions and further depolarisation of the membrane potential towards the equilibrium potential for sodium (~+60 mV in most neurons). VGSCs close within milliseconds of opening. Inactivation of VGSCs is usually incomplete, resulting in a small persistent Na+ current, which inactivates over a time period of tens of seconds. This can have important functional consequences (e.g. Braho et al., 2016). VGSCs can be divided into two parts with the transmembrane domains S1–S4 contributing to the voltage sensor and S5–S6 arranging to form the sodium selective pore. The VGSC inactivation gate contains three amino acids (isoleucine, phenylalanine and methionine (IFM)) located in the intracellular loop connecting domains III and IV. Progress has been made in determining the structures of sodium channels using cryoelectron microscopy and X-ray crystallography (Catterall, 2014; Clairfeuille et al., 2016). However, the conformations present in the multi-molecular structures found in a neuronal membrane are likely to be variants on these basic structures.
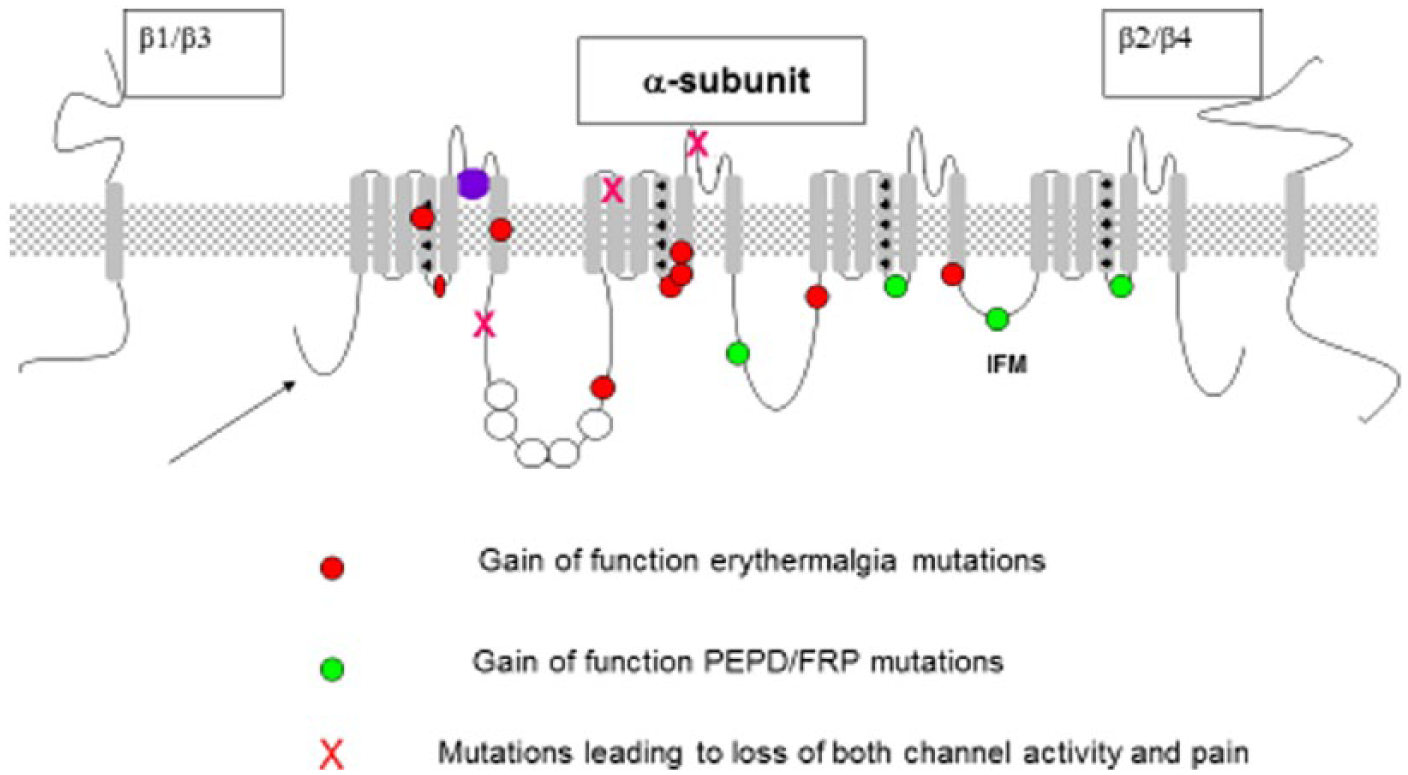
Voltage-gated sodium channels – human NaV1.7 pain-related mutations (Emery et al. 2016).
Sodium channels are invariably associated with accessory β-subunits as well as other proteins that may play a role in anchoring the functional sodium selective pore in specific cellular locations. Most interest has focused on the mechanism that associates voltage-gated channels like NaV1.6 with nodes of Ranvier, to facilitate fast saltatory conduction, a topic that has recently been reviewed (Freeman et al., 2016). Below we discuss a variety of human pathologies that have been linked to sodium channel mutations.
Sodium channels and epilepsy
Broadly expressed in the central nervous system (CNS), NaV1.1 expression is found in inhibitory GABAergic neurons. The majority of the SCN1A mutations causing the dominant epilepsy disorder Dravet’s syndrome are nonsense mutations. In addition, SCN1A mutations have been identified in families with the milder disorder, generalised epilepsy with febrile seizures, which is characterised by short-lasting tonic–clonic seizures accompanied by fever. Generalised epilepsy with febrile seizure mutations changes the expression and function of NaV1.1 channels due to both gain- and loss-of-function mutations.
The more detrimental Dravet’s syndrome is associated with haploinsufficiency for SCN1A in 50%–80% of severe myoclonic epilepsy of infancy patients caused by more deleterious nonsense and frameshift mutations in NaV1.1. In contrast to generalised epilepsy with febrile seizures plus, these mutations prevent channel expression or severely impair the channel function. While loss-of-function mutations are common in Dravet’s syndrome, a gain-of-function mutation in SCN1A (R865G) has also been found. SCN1A duplications and deletions are also found in patients with Dravet’s syndrome.
The severity of channel impairment has been suggested to underlie the different efficacies of some anti-epileptic drugs in treating either generalised epilepsy with febrile seizures plus or severe myoclonic epilepsy of infancy. For example, the sodium channel blocker lamotrigine is very effective for treating generalised epilepsy with febrile seizures plus, while it aggravates symptoms in patients with severe myoclonic epilepsy of infancy. Studies on Scn1a+/− mice have shown that the deletion of NaV1.1 leads to impaired firing of GABAergic inhibitory hippocampal interneurons and cerebellar GABAergic Purkinje neurons. The impaired functioning of inhibitory GABAergic neurons may contribute to seizures, ataxia, spasticity and failure of motor coordination observed in these mice. NaV1.1 mutations are also associated with familial hemiplegic migraine type 3, an autosomal dominant severe subtype of migraine with aura characterised by hemiparesis during the attacks. Whole exome sequencing has also identified candidate genes with de novo mutations, including SCN1A, in sporadic autism spectrum disorders.
NaV1.2 is abundantly expressed, in the cortex and hippocampus in regions destined to become nodes of Ranvier where it is replaced during development by NaV1.6. NaV1.2 knockout mice die perinatally from neuronal apoptosis and hypoxia. In humans, NaV1.2 mutations are associated with inherited epilepsy, mainly benign familial neonatal–infantile seizures and less frequently generalised epilepsy with febrile seizures. Two independent nonsense mutations in SCN2A have also been linked to autism (Eijkelkamp et al., 2012).
Sodium channels, muscle and cardiac function
NaV1.4 is responsible for the generation and propagation of action potentials that initiate muscle contraction. Hereditary sodium channelopathies of skeletal muscle involving NaV1.4 mutations have been identified, such as hyperkalaemic periodic paralysis, hypokalaemic periodic paralysis, paramyotonia congenita and congenital myasthenic syndrome. Hypokalaemic periodic paralysis and normokalaemic periodic paralysis mutations map to the NaV1.4 voltage sensor, resulting in sodium leak through the gating pore allowing sustained inward sodium flux at negative membrane potentials.
Unlike the skeletal muscle channel NaV1.4, the cardiac channel NaV5 has been detected in a number of neuronal subtypes as well including sensory neurons. Several syndromes leading to sudden cardiac death have been linked to mutations in NaV1.5. Brugada syndrome leads to sudden cardiac death that may account for up to 50% of all sudden cardiac deaths in young individuals without structural heart disease. SCN5A mutations were found in ~20% of patients with Brugada syndrome resulting in channel loss of function through a number of different mechanisms including the expression of non-functional NaV1.5, decreased protein expression or impaired membrane trafficking. Interestingly, VGSC upregulation has been associated with several strongly metastatic carcinomas, leading to the hypothesis that VGSC upregulation may ‘switch’ the cancerous cell to an invasive state. Some cancers express embryonic/neonatal VGSC splice variants, for example, a neonatal isoform of NaV1.5 (seven amino acid differences) is the predominant (>80%) VGSC in human metastatic breast cancer as well as neuroblastoma. More recently, the intriguing observation has been made that mutations in non-coding regions of NaV1.8 may play an important role in the regulation of the expression of NaV1.5 and Brugada syndrome (Hu et al., 2014). NaV1.8 is not expressed within cardiac muscle, but is nonetheless an important risk factor for heart disease lying adjacent to NaV1.5 in the genome.
Sodium channels and pain
The fact that sodium channel blockers are effective analgesics has focussed attention on the three sensory neuron isoforms NaV1.7–NaV1.9 mainly found in the peripheral nervous system (Waxman et al., 2014). In fact, only NaV1.8 is highly selectively expressed in sensory neurons, while NaV1.7 and NaV1.9 are found in a variety of CNS neurons as well.
NaV1.8 is expressed in nociceptive sensory neurons (Akopian et al., 1999) and acts as a major contributor to the upstroke of action potentials. NaV1.8 is essential in maintaining the excitability of nociceptors at low temperatures, becoming the sole electrical impulse generator at temperatures <10°C. This is caused by enhanced slow inactivation of TTX-sensitive channels in response to cooling, whereas the inactivation of NaV1.8 is cold resistant. Antisense oligonucleotides attenuate the development and maintenance of neuropathic pain, while small interfering RNA selective knockdown of NaV1.8 reverses mechanical allodynia. However, NaV1.8 knockout mice show normal neuropathic pain behaviour. However, selective blockers of NaV1.8 successfully suppress various pain symptoms and neuropathic pain in rats. A recent genome-wide association study has identified a single-nucleotide polymorphism in NaV1.8 which was associated with prolonged cardiac conduction (longer P-wave duration), thereby providing evidence that NaV1.8 has a functional role in the heart through regulation of the expression of the cardiac channel NaV1.5.
NaV1.9 is the most recently discovered sodium channel subtype. It is a marker of primary nociceptors and is also expressed in the enteric nervous system and motor neurons. NaV1.9 is a biophysically unique sodium channel which generates TTX-resistant currents that have very slow gating kinetics. The current generated by NaV1.9 is ‘persistent’ and can be activated at potentials close to resting membrane potential (~−60 mV), and the channel acts as a modulator of membrane excitability by contributing regenerative inward currents over a strategic membrane potential range both negative to and overlapping with the voltage threshold for other transient sodium channels.
While a selective blocker of NaV1.9 does not exist at present, SCN11A knockout mice exhibit a clear analgesic phenotype, confirming that NaV1.9 is an important player in generating hyperalgesia in inflammatory pain states. This appears to be explicable by changes in the properties of distal primary afferents. The response to inflammatory mediators is suppressed in NaV1.9 knockout mice consistent with the immunocytochemical localisation of the channel at unmyelinated nerve endings, and the remarkable functional plasticity of the current, known to be under G-protein pathway control via protein kinase C. Targeting NaV1.9 could help regulate pain thresholds following inflammation or injury.
A number of human heritable pain disorders map to mutations in SCN9A, the gene encoding NaV1.7. Dominant gain-of-function mutations lead to inherited primary erythromelalgia, which is characterised by bilateral burning pain of the feet/lower legs and hands, elevated skin temperature of the affected areas and reddened extremities. In addition, dominant gain-of-function mutations can cause paroxysmal extreme pain disorder which is characterised by episodic burning pain of the rectum, ocular and mandibular regions. Rarer recessive loss-of-function conditions cause an inability to experience pain and anosmia (Waxman et al., 2014).
In mice in which NaV1.7 is deleted from all sensory neurons as well as sympathetic neurons, there is an almost complete loss of pain sensation, consistent with human studies (Minett et al., 2015). Overall, the essential role of NaV1.7 in human as well as animal pain perception highlights NaV1.7 as an important analgesic drug target.
It is thus all the more puzzling that truly selective NaV1.7 antagonists have no analgesic activity whatsoever when applied to the periphery (Deuis et al., 2017). Reports of NaV1.7 neutralising monoclonal antibodies or specific toxins with analgesic effects have proved impossible to repeat. The reason for this anomaly is that loss of NaV1.7 upregulates the expression of both opioid peptides and the activity of their cognate G protein-coupled receptors (GPCRs). In order to effect these actions, a complete loss of NaV1.7 function is required – something that is pharmacologically almost impossible to achieve. Thus, the opioid system, upregulated in mice and men that lack NaV1.7, contributes to a pain-free state over a whole lifetime, implying that manipulating the endogenous opioid system could in principal give us analgesia without tolerance or addiction of other unpleasant side effects – a very enticing prospect (Emery et al., 2016).
The mechanisms linking sodium channel activity to these effects on opioid signalling are of great mechanistic significance in terms of developing new routes to analgesia. Intriguingly, NaV1.7 has been shown to be necessary for neurotransmitter release from olfactory sensory neurons, as well as playing an unsuspected role in the hypothalamus, integrating synaptic inputs over long periods to regulate the release of hormones involved in regulating body weight (Branco et al., 2016). NaV1.7’s fascinating roles in a range of physiological processes far removed from simple action potential propagation suggest that more surprises in terms of sodium channel function will become apparent in the coming years.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors gratefully acknowledge financial support from the Wellcome Trust and Astra Zeneca.