
Abstract
Background:
Nuclear factor I family members nuclear factor I A and nuclear factor I B play important roles during cerebral cortical development. Nuclear factor I A and nuclear factor I B regulate similar biological processes, as their expression patterns, regulation of target genes and individual knockout phenotypes overlap. We hypothesised that the combined allelic loss of Nfia and Nfib would culminate in more severe defects in the cerebral cortex than loss of a single member.
Methods:
We combined immunofluorescence, co-immunoprecipitation, gene expression analysis and immunohistochemistry on knockout mouse models to investigate whether nuclear factor I A and nuclear factor I B function similarly and whether increasing allelic loss of Nfia and Nfib caused a more severe phenotype.
Results:
We determined that the biological functions of nuclear factor I A and nuclear factor I B overlap during early cortical development. These proteins are co-expressed and can form heterodimers in vivo. Differentially regulated genes that are shared between Nfia and Nfib knockout mice are highly enriched for nuclear factor I binding sites in their promoters and are associated with neurodevelopment. We found that compound heterozygous deletion of both genes resulted in a cortical phenotype similar to that of single homozygous Nfia or Nfib knockout embryos. This was characterised by retention of the interhemispheric fissure, dysgenesis of the corpus callosum and a malformed dentate gyrus. Double homozygous knockout of Nfia and Nfib resulted in a more severe phenotype, with increased ventricular enlargement and decreased numbers of differentiated glia and neurons.
Conclusion:
In the developing cerebral cortex, nuclear factor I A and nuclear factor I B share similar biological functions and function additively, as the combined allelic loss of these genes directly correlates with the severity of the developmental brain phenotype.
Keywords
Introduction
The nuclear factor I (NFI) family of transcription factors play major roles during embryogenesis. Nuclear factor I-A (NFIA) and nuclear factor I-B (NFIB) are important for normal cortical development, as knockout of either Nfia or Nfib in mice results in very similar brain phenotypes, including dysgenesis of the corpus callosum, the disruption of midline fusion, enlarged ventricles and malformation of the hippocampus (Das Neves et al., 1999; Gobius et al., 2016; Piper et al., 2010; Shu et al., 2003; Steele-Perkins et al., 2005). Similarly, mutations or deletions of NFIA and NFIB in human patients with intellectual disability are associated with severe brain phenotypes, including dysgenesis of the corpus callosum, macrocephaly and enlarged ventricles (Gobius et al., 2016; Koehler et al., 2010; Lu et al., 2007; Negishi et al., 2015; Sajan et al., 2013).
Analyses of knockout mouse models suggest that the general underlying cause of these phenotypes is a cellular differentiation defect which affects radial glia, the embryonic neural progenitor cells of the cerebral cortex, which give rise to mature neurons and glia in the cortex (Barry et al., 2008; Betancourt et al., 2014; Gobius et al., 2016; Piper et al., 2009, 2010, 2014). Knockout of either Nfia or Nfib causes the radial glia to remain proliferative and undifferentiated for an extended period of time. As a result, the generation of differentiated cellular progeny in the cortex is delayed (Barry et al., 2008; Gobius et al., 2016; Shu et al., 2003), a phenotype that is also observed in the cerebellum and the spinal cord (Deneen et al., 2006; Glasgow et al., 2013; Kilpatrick et al., 2012).
NFIA and NFIB proteins share highly homologous DNA-binding domains (Gronostajski, 2000) and have nearly identical DNA-binding motifs (Jolma et al., 2013). In the context of the brain, NFIA and NFIB share many common transcriptional downstream targets, including Gfap, Hes1, Hes5, Sox3, Fgf9, Gli3, Id4, Cntn2, Efnb1 and Cdh2 (Barry et al., 2008; Betancourt et al., 2014; Brun et al., 2009; Cebolla and Vallejo, 2006; Harris et al., 2016; Piper et al., 2009, 2010, 2014; Wang et al., 2007, 2010). In vitro experiments for targets such as Gfap and Cntn2 have validated their direct regulation by both NFIA and NFIB. In contrast, transcriptional targets identified outside the central nervous system, such as IGFBP5, PPARγ, C/EBPα, FABP4, BRN2 and NKX3.1 (Fane et al., 2017; Grabowska et al., 2014; Perez-Casellas et al., 2009; Waki et al., 2011), are differentially regulated by NFIA and NFIB. Hence, specifically within the developing brain, NFIA and NFIB may share a conserved biological function.
In this study, we investigated whether NFIA and NFIB regulate similar biological processes during early cerebral development, as suggested by the overlapping phenotypes observed in mouse models and human patients. This would imply that within this context, NFIA and NFIB could function, at least in part, additively, and that the total ‘gene dosage’ of both factors could be important for normal cortical development. From these lines of evidence, we hypothesised that combined knockout of one allele of both Nfia and Nfib would result in a similar phenotype to the biallelic knockout of either family member, whereas knockout of more alleles of these genes would exacerbate the dysregulation of cellular differentiation and the observed brain developmental phenotype.
Materials and methods
Animal breeding
All breeding and experiments were performed at the State University of Buffalo under approval from the Institutional Animal Care and Use Committee, or at the University of Queensland in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and with approval from the University of Queensland Animal Ethics Committee. Nfia knockout (Nfiatm1Rmg) (Das Neves et al., 1999), Nfib knockout (Nfibtm1Rmg) (Steele-Perkins et al., 2005), Nfibcond conditional knockout (Nfibtm2Rmg) (Hsu et al., 2011) and R26-CreERT2 (Gt(ROSA)26Sortm1(Cre/ERT2)Tyj) (Ventura et al., 2007) mice were maintained on a C57Bl/6 background. Individual alleles were genotyped as described previously (Das Neves et al., 1999; Harris et al., 2016; Hsu et al., 2011; Steele-Perkins et al., 2005).
To generate timed-pregnant females, male and female mice were placed together overnight, and the females checked the following day for vaginal plugs. This day was designated as embryonic day (E)0 if a vaginal plug was present. To induce deletion of the Nfibcond allele in Nfia;Nfibcond;Rosa26-CreERT mice, 2 mg tamoxifen (T-5648; Sigma) in corn oil was administered by intraperitoneal injection into dams at E10.5, E11.5 and E12.5 to generate double heterozygous (Nfia+/−;Nfib+/−) and homozygous (Nfia–/−;Nfib−/−) knockout embryos. Dams were euthanised using sodium pentobarbital (Abbott Laboratories), and embryos were drop fixed (E14 and below) or transcardially perfused with 0.9% (w/w) saline, followed by 4% paraformaldehyde in phosphate-buffered saline (PBS; pH 7.4) for further immunohistochemical analyses. For co-immunoprecipitation and RNA isolation, neocortical tissue was microdissected in ice-cold PBS. Samples used for RNA isolation were immediately snap frozen on dry ice, whereas co-immunoprecipitation was performed with fresh tissue samples. Frozen tissue was stored at −80°C until RNA isolation (Bunt et al., 2015).
RNA sequencing and analysis
Total RNA was isolated from frozen neocortical tissue using TRIzol Reagent as per the manufacturer’s protocol (Life Technologies). Isolated RNA was treated with DNase (DNA-free DNA removal kit; Life Technologies). Libraries were prepared for 2 wildtype and 4 knockout Nfia−/− and 2 wildtype and 4 knockout Nfib−/− samples using the TruSeq RNA Library Preparation Kit v3 (paired-end 100nt length) and sequenced on an Illumina HiSeq 2000 (Illumina).
For analysis, paired-end reads from each sample were aligned to the mm9 reference genome using the TopHat2 algorithm version 2.0.10 (Kim et al., 2013). The change in expression in the Nfia and Nfib knockout tissue was calculated using gene expression levels in four Nfia and four Nfib knockout samples and their respective wildtype littermate samples (GSE93604). The fold change in expression and statistical significance were calculated using the CuffDiff tool version 2.1.1 (Trapnell et al., 2010).
Genes were considered regulated if the 2log fold change of expression was >0.4 and the q-value was <0.05. Gene ontology analyses were performed using the DAVID tool (Huang da et al., 2009a, 2009b). To predict the presence of NFI binding motifs, we used the Find Individual Motif Occurrences tool (FIMO; Grant et al., 2011) with default programme significance threshold and the NFI motifs defined by Jolma et al. (2013). Enrichment of NFIB binding peaks (Chang et al., 2013; Lajoie et al., 2014) in the promoter were analysed as described previously (Bunt et al., 2012) and significance was determined by a chi-square test with Yates’ corrections.
Real-time quantitative polymerase chain reaction analyses
Reverse transcription and real-time quantitative polymerase chain reaction (qPCR) analyses were performed on messenger RNA (mRNA) isolated from E16 Nfia−/−, Nfib−/− or wildtype littermates as described previously (Lim et al., 2015). The thermocycler conditions used were as follows: 2 min at 50°C and 10 min at 95°C, followed by 45 cycles with 10 s denaturation at 95°C, 15 s annealing at 60°C and 20 s extension at 72°C. Relative expression was determined using the ΔΔCt method with Hprt as a reference gene. Statistical significance was determined using two-tailed Student’s t-tests. The primer sequences were as follows: Alcam, 5′-GGCAGTGGGAGCGTCATAAAC-3′ and 5′-ATCGCAGAGACATTCAGGGAG-3′; Hprt, 5′-GCAGTACAGCCCCAAAATGG-3′ and 5′-AACAAAGTCTGGCCTGTATCCAA-3′; Id4, 5′-CAGTGCGATATGAACGACTGC-3′ and 5′-GACTTTCTTGTTGGGCGGGAT-3′; Igfbp3, 5′-CCAGGAAACATCAGTGAGTCC-3′ and 5′-GGATGGAACTTGGAATCGGTCA-3′; Mycn, 5′-AACAACAAGGCGGTAACCAC-3′ and 5′-GAGGGTGCAGCATAGTTGTG-3′; Pou3f1, 5′-TCGAGGTGGGTGTCAAAGG-3′ and 5′-GGCGCATAAACGTCGTCCA-3′; Tle4, 5′-TTTACAGGCTCAATACCACAGTC-3′ and 5′-TGCACAGATAGCATTTAGTCGTT-3′.
Co-immunoprecipitation
Cytoplasmic and nuclear lysates were prepared from dissected neocortical tissue from E13 C57Bl/6J wildtype embryos as described previously (Klenova et al., 2002). A volume of 5 μg of co-immunoprecipitation antibody was pre-incubated with 15 μL Pierce Protein G magnetic beads (Thermo Scientific) in Tris-buffered saline containing 0.05% Tween-20 (v/v) (TBS.T; pH 7.5) for 4 h at 4°C prior to co-immunoprecipitation. Antibodies used for co-immunoprecipitation were rabbit anti-NFIA (HPA008884; Sigma), rabbit anti-NFIB (HPA003956; Sigma) and normal rabbit IgG (2729; Cell Signaling Technology). Antibody-bound beads were washed with TBS.T and co-immunoprecipitation buffer, and then incubated with nuclear lysate for 16 h at 4°C. Following co-immunoprecipitation, antibody-bound beads were washed thrice with co-immunoprecipitation buffer and prepared for Western blotting with NuPAGE LDS Sample Buffer (Invitrogen) and 0.1 M DL-dithiothreitol (Sigma). Samples were denatured by heating to 85°C for 10 min and electrophoresed on NuPAGE Novex 4%–12% Bis-Tris gels (Invitrogen). Electrophoresed samples were transferred to an Immobilon-FL polyvinylidene difluoride (PVDF) membrane (Merck Millipore) using the XCell II Blot Module (Invitrogen) according to the manufacturer’s recommendations. Blotted membranes were blocked with 5% (w/v) skim milk powder in PBS and incubated with the appropriate primary antibody diluted with 5% (w/v) skim milk powder in PBS containing 0.1% (v/v) Tween-20 (PBS.T). The primary antibodies used for Western blots were rabbit anti-NFIA (1:1000, 39,397; Active Motif) and mouse anti-NFIB (1:100, ab51352; Abcam). The secondary antibodies used were IRDye 680LT donkey anti-rabbit and IRDye 800CW donkey anti-mouse (both diluted 1:15,000; LI-COR) diluted in PBS.T. Membranes were imaged using the Odyssey Classic (LI-COR) and Image Studio 5 software (LI-COR).
Immunohistochemistry and immunofluorescence
Dissected and post-fixed brains were embedded in 3% noble agar (Difco Sparks), and 50 μm coronal sections were cut using a vibratome (Lecia). Free-floating staining was performed as described previously (Barry et al., 2008; Campbell et al., 2008). The primary antibodies used for immunohistochemistry were rabbit anti-GFAP (1:30,000, Z0334; Dako), mouse anti-GAP43 monoclonal antibody (1:50,000, MAB1987; Millipore) and rabbit anti-TBR1 polyclonal antibody (1:1000, sc48816; Santa Cruz Biotechnology). A biotinylated goat anti-rabbit or anti-mouse secondary antibody (1:500; Jackson ImmunoResearch) was used, followed by incubation with the VECTASTAIN elite ABC kit (Vector Laboratories) and 3,3′-diaminobenzidine (DAB) staining as previously described (Plachez et al., 2008). Immunofluorescence co-staining on mounted sections was performed as described previously (Plachez et al., 2008). Sections were incubated with rabbit anti-NFIA (1:500, HPA008884; Sigma), rabbit anti-NFIB (1:500, HPA003956; Sigma), chicken anti-ß-galactosidase (1:500, ab9361; Abcam), rabbit anti-TBR1 polyclonal antibody (1:1000, sc48816; Santa Cruz Biotechnology), rat anti-CTIP2 (1:500, ab18465; Abcam), mouse anti-NeuN (1:1000, MAB377; Chemicon) followed by donkey Alexa Fluor 488, 555 or biotinylated labelled secondary antibodies (Invitrogen) and Alexa Fluor 647-conjugated Streptavidin (Invitrogen) amplification . Cell nuclei were stained using haematoxylin (Sigma) or 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). For all genotypes, matching sections of a minimum of six animals were analysed, except for Nfia;Nfib double homozygous knockout mice, for which three embryos were analysed due to the reduced viability of these embryos.
Imaging and analyses
Brightfield imaging was performed with a Zeiss upright Axio-Imager Z1 microscope fitted with Axio-Cam HRc and HRm cameras. Images were acquired with the ZEN blue software (Carl Zeiss). Immunofluorescence images were obtained from a Diskovery inverted spinning-disk confocal system (Spectral Applied Research) consisting of a Ti-E microscope (Nikon) equipped with a Diskovery disk head (Spectral Applied Research), two Flash4.0 sCMOS cameras (Hamamatsu Photonics) and 20× 0.75 NA CFI PlanApo and 40× 1.15 NA CFI ApoLambda objectives. Images were pseudo-coloured to permit overlay and were then cropped, sized and contrast-brightness enhanced for presentation with Photoshop software (Adobe). For quantification, cortical measurements were taken from haematoxylin-stained matched sections of three to five animals per condition. All measurements were averaged from both hemispheres of each animal. Statistical significance was determined using two-tailed Student’s t-test.
Results
Radial glia co-express NFIA and NFIB during development
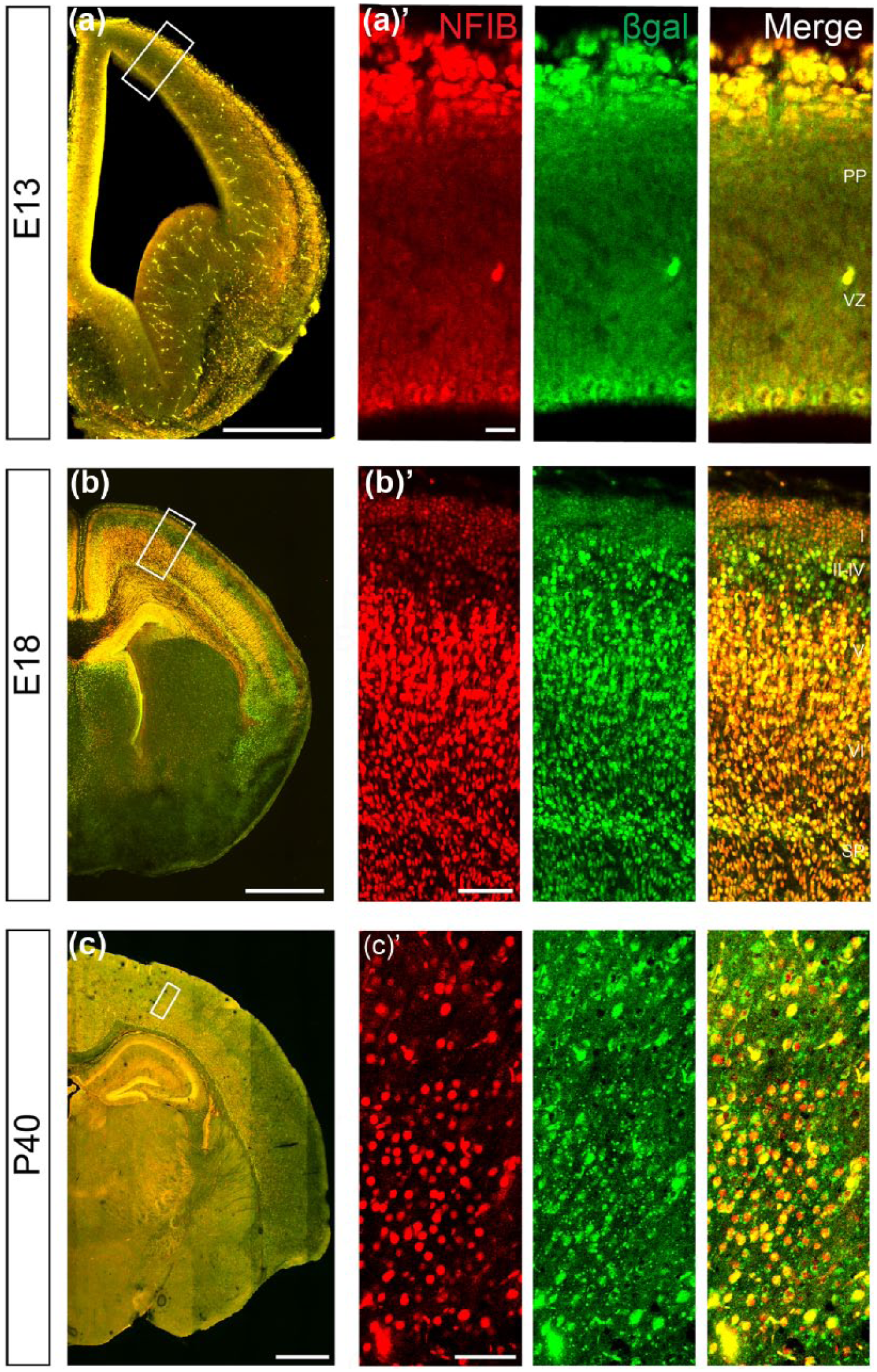
The individual expression of NFIA and NFIB has been reported in radial glia from E12 in mice (Betancourt et al., 2014; Bunt et al., 2015; Plachez et al., 2008), but their co-expression within these cells has not been determined. To exclude the possibility that knockout of Nfia and Nfib affected different subpopulations of radial glia, we analysed their co-expression at a cellular level in the developing cerebrum. The Nfib locus carries a knock-in β-galactosidase reporter gene substituted into the deleted exon of the Nfib allele as a marker for NFIB expression (Betancourt et al., 2014; Piper et al., 2009; Steele-Perkins et al., 2005). For each stage examined, we validated β-galactosidase and NFIB co-expression (Figure 1) to ensure that we were able to analyse co-expression of NFIA and NFIB using β-galactosidase protein expression as a reporter with fidelity (Figure 2). At E13, both NFIA and β-galactosidase were co-expressed in a high medial to low lateral gradient in the coronal plane throughout the ventricular zone (VZ) of the cerebral cortex. At later stages of development, such as E18, the VZ of the cerebral cortex and hippocampus still co-expressed both proteins, but their expression had started to diverge in the post-mitotic progeny (Figure 2(b)–(e) and Figure 3). While NFIA and β-galactosidase were co-expressed in the subplate and layer VI of the neocortex, NFIB expression extended more dorsally than NFIA at E18 (Figures 2(b) and 3). Similarly, expression of β-galactosidase extends further into the hippocampal plate. Postnatally, NFIA and NFIB remained co-expressed in the neural progenitor cells of the dentate gyrus and by the ependymal cells lining the lateral ventricles (Figure 2(c) and (e)). In line with our previous work detailing expression of NFI proteins in the adult mouse brain (Chen et al., 2017), NFIA and NFIB remained highly co-expressed in the pyramidal layer and dentate gyrus in the hippocampus, while the expression of these factors within the neocortex was more diverse. Hence, deletion of Nfia and Nfib will likely affect the same population of radial glia.


NFIA and NFIB share regulation of downstream target genes involved in neurodevelopment
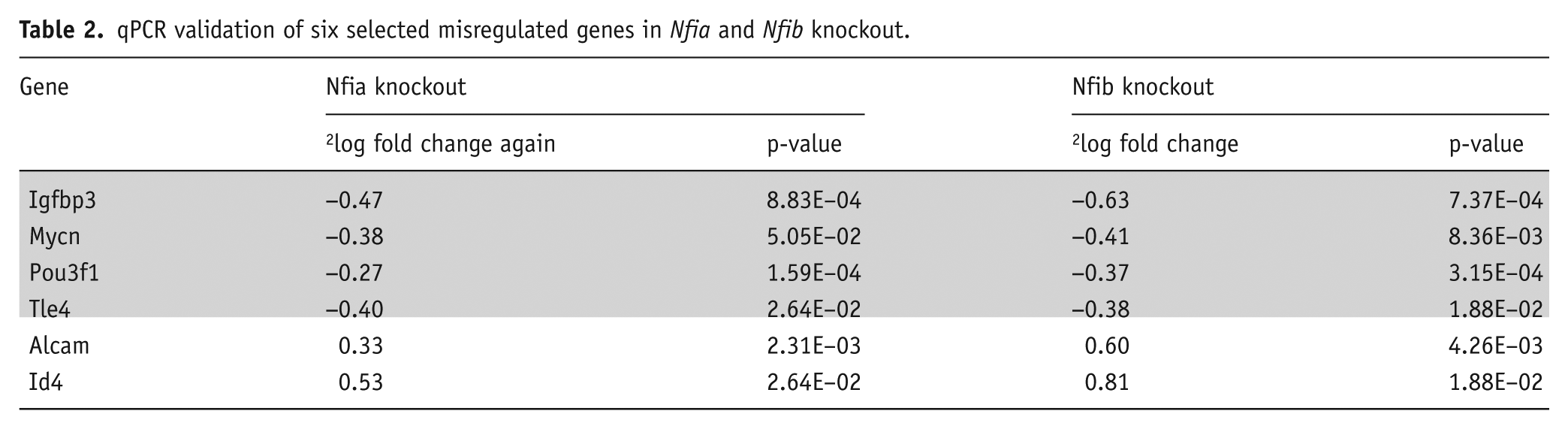
Although mRNA expression analyses of Nfia and Nfib knockout embryos have revealed several common downstream targets (Barry et al., 2008; Betancourt et al., 2014; Harris et al., 2016; Piper et al., 2009, 2010, 2014), the overlap of these factors in regulating downstream targets and pathways remained unclear. We therefore analysed mRNA-sequencing data from the cerebral cortex of E16 Nfia and Nfib knockout mice and their corresponding wildtype littermates. Using thresholds of 2log fold change >0.4 and q-value <0.05, 540 and 738 genes were differentially regulated in Nfia and Nfib knockout mice, respectively. Of these, 91 genes showed similar misregulation in both Nfia and Nfib knockout mice, when we allowed for a less stringent q-value of <0.1 in one mouse model to compensate for experimental variability (Table 1). We validated the misregulation of the genes Alcam, Id4, Igfbp3, Mycn, Pou3f1 and Tle4 by qPCR (Table 2).
Common misregulated genes in Nfia and Nfib knockout E16 neocortical tissue.

qPCR validated.
qPCR validation of six selected misregulated genes in Nfia and Nfib knockout.
Functional annotation clustering using DAVID (Huang da et al., 2009a, 2009b) confirmed that the 91 shared genes were enriched for neurodevelopment-related gene ontology terms (Table 3; p < 0.05 with Benjamini–Hochberg correction). Similarly, when we performed gene ontology separately on the differentially regulated genes identified from only one mouse model (Table 4; 540 genes for Nfia and 738 genes for Nfib; p < 0.005 with Benjamini–Hochberg correction), the gene ontology terms that were common to both mouse models also consisted of neurodevelopment-related processes. In contrast, gene ontology terms unique to only one Nfi knockout model were also related to NFI function in other organs, such as lung development for NFIB (Steele-Perkins et al., 2005). Collectively, these results suggest that NFIA and NFIB are likely to co-regulate a core neurodevelopmental programme to drive embryonic corticogenesis.
Gene ontology enrichment of 91 common misregulated genes in Nfia and Nfib knockout E16 neocortical tissue.

Gene ontology enrichment for biological processes of regulated targets in Nfi knockout E16 cortex.

Shared misregulated genes in Nfi knockout neocortex are enriched for NFI binding
To test whether the 91 shared misregulated genes are more likely to represent direct NFI targets, we determined whether predicted NFI binding motifs (Jolma et al., 2013) were present in their respective promoters. Although such a motif was present in 59.5% of all genes in the genome within 2000 base pairs of the transcription start sites, this value increased to 71.7% and 71.3% in the 540 and 738 differentially regulated genes identified in Nfia and Nfib knockout mice, respectively (Figure 4(a)). The 91 similarly misregulated genes showed even higher enrichment, with 85.7% having at least one predicted NFI binding motif.

Not all predicted binding motifs might represent true NFI binding sites. To assess whether NFIB could bind to the promoter of these differentially expressed genes, we analysed the presence of an NFIB binding peak in their promoter using published NFIB chromatin immunoprecipitation-sequencing (ChIP-sequencing) data reported in other tissues (Chang et al., 2013; Lajoie et al., 2014). The presence of an NFIB binding peak detected within a gene promoter from a ChIP-sequencing dataset from mouse hair follicle stem cells increased from 1.1% in the promoter of all genes to 1.5% and 2.7% in the differentially regulated genes that we identified in the Nfia and Nfib knockout mice, respectively (Figure 4(b)). Similarly, the presence of an NFIB binding peak detected within a ChIP-sequencing dataset from E16 lung tissue increased from 6.9% (all genes) to 10.2% (Nfia knockout) and 12.6% (Nfib knockout) (Figure 4(c)). The misregulated genes in our analysis were therefore more likely to contain a putative NFI binding site within their promoter.
The 91 misregulated genes shared between Nfia and Nfib showed even higher enrichment for putative NFI binding sites: 5.5% and 17.6% in hair follicle and lung datasets, respectively (Chang et al., 2013; Lajoie et al., 2014). NFIB binding peaks were more prevalent in the genes that were down-regulated following Nfi deletion (striped bars in Figure 4(b) and (c)), displaying enrichments of 8.1% (hair follicle) and 22.6% (lung). This enrichment increased to 9.8% and 34.1%, respectively, when we increased the minimal 2log fold change to >0.6. Hence, NFI-activated genes that are down-regulated in both Nfia and Nfib knockout mice are more likely to represent direct targets of both NFIA and NFIB.
To identify shared targets that are expressed specifically within cortical radial glia, we utilised the differential expression modules described for the E14.5 neocortex by Fietz et al. (2012). Compared to all genes (8.1%), genes that were down-regulated in both Nfia and Nfib knockout mice were enriched for high expression in the VZ (23.1%, or 12 out of 52 shared down-regulated genes; Yates < .0001) (Tables 1 and 2). We were also able to verify the high VZ expression of these 12 genes using in situ hybridisation datasets (Tables 1 and 2) (Allen Institute, 2013; Diez-Roux et al., 2011; Kawaguchi et al., 2008; Shimogori et al., 2010; Visel et al., 2004). Furthermore, 7 out of the 12 down-regulated genes showed NFIB binding in their promoter in either lung or hair follicle ChIP-sequencing datasets, while only 9 out of the remaining 40 down-regulated genes had binding. Hence, these seven genes, Bcan, Kcne1l, Loxl1, Ltbp1, Mfge8, Slc9a3r1 and Tnc, are likely to be directly regulated by NFIA and NFIB in radial glia, and may be responsible for the similar cortical developmental defects that are observed in single knockout mice.
NFIA and NFIB dimerise in vivo
Besides recognising the same DNA motif (Jolma et al., 2013), NFIA and NFIB could physically interact with each other through protein dimerisation. The NFI DNA binding motif consists of two palindromic recognition sites, and in vitro analysis has demonstrated the binding of two NFI proteins at such sites (Gronostajski et al., 1985; Leegwater et al., 1985). Furthermore, both hetero- and homodimers have been observed in vitro (Kruse and Sippel, 1994). However, whether heterodimerisation occurs in vivo remains unknown. To investigate whether NFIA and NFIB physically interact in the developing cortex, we performed co-immunoprecipitation of NFIA and NFIB using E13 neocortical lysates (Figure 4(d)). Compared to the input, NFIA and NFIB proteins were enriched in the immunoprecipitate, obtained using antibodies that specifically recognise the other family member, thereby demonstrating that heterodimerisation can occur in vivo. Hence, NFIA and NFIB can potentially heterodimerise to co-regulate their target gene expression.
Double heterozygous Nfia;Nfib knockout embryos have similar brain defects to those of single homozygous knockout mice
To test whether the total number of Nfi alleles modulates cortical development, we crossed Nfia knockout (Das Neves et al., 1999) with Nfib conditional (Hsu et al., 2011); R26-CreERT (Ventura et al., 2007) mice. Tamoxifen injection at embryonic day E10.5, E11.5 and E12.5 generated Nfia;Nfib double heterozygous knockout embryos (hereafter referred to as Nfia+/−;Nfib+/−). At E18, all Nfia+/−;Nfib+/− embryos displayed retention of the interhemispheric fissure and dysgenesis of the corpus callosum as shown by axonal GAP43 staining (Figure 5(a)–(h)). Furthermore, the hippocampi of these embryos showed a distinct malformation, with a reduction in the dentate gyrus, again comparable to the individual null strains (Figure 5(i)–(l)). Hence, the phenotype of the Nfia+/−;Nfib+/− embryos was very similar to that of Nfia−/− or Nfib−/− single knockout embryos (Barry et al., 2008; Das Neves et al., 1999; Piper et al., 2009, 2010, 2014; Shu et al., 2003; Steele-Perkins et al., 2005).
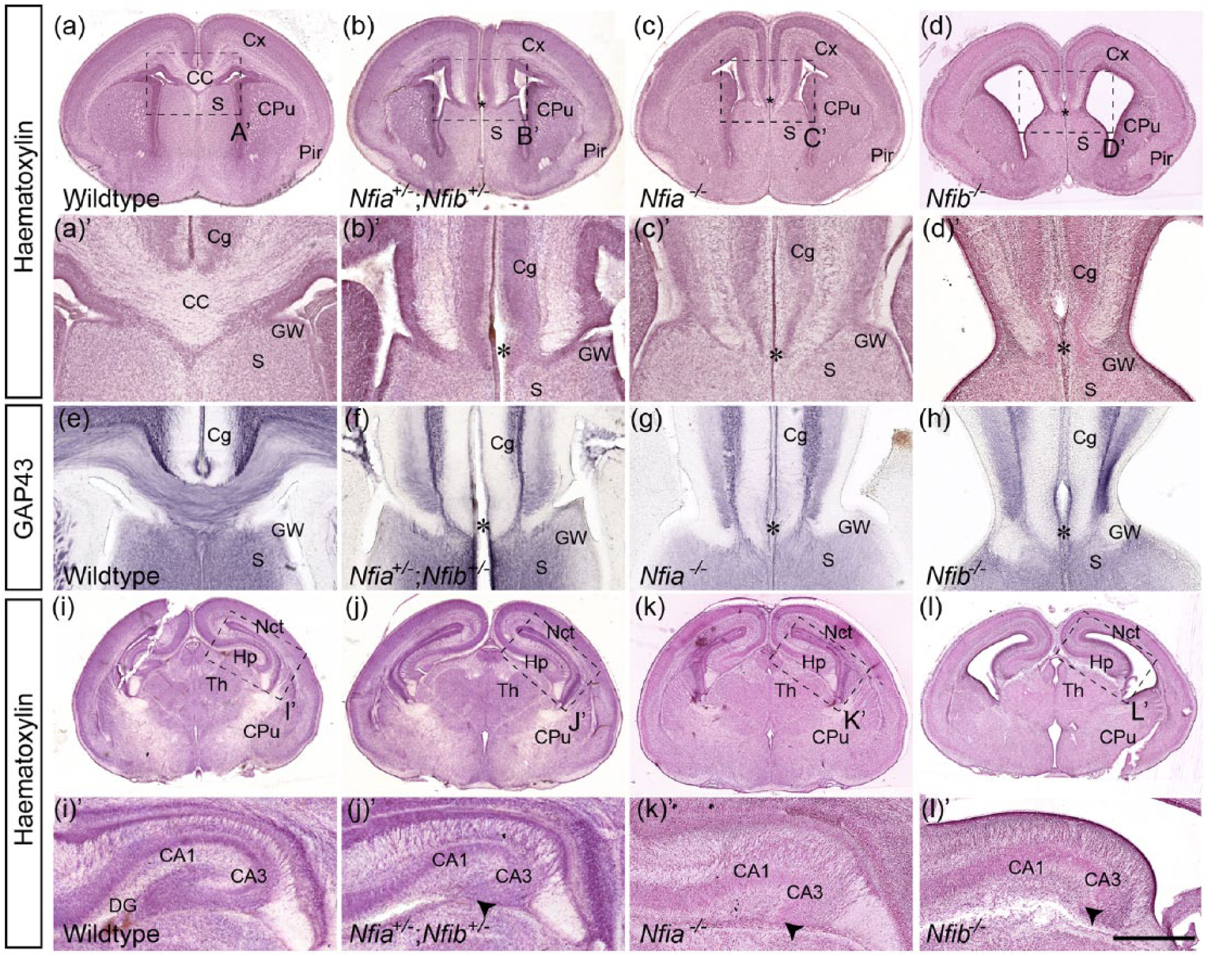
On a cellular level, the brains of Nfia+/−;Nfib+/− mice displayed a reduction in astroglial cells as assessed by the expression of the mature astroglial marker GFAP, a finding consistent with that observed in single homozygous knockout embryos (Figure 6) (Shu et al., 2003; Steele-Perkins et al., 2005). At the midline, double heterozygous brains showed a reduction in all three glial populations. GFAP expression was completely absent in the glial wedge, as was observed in single Nfia−/− or Nfib−/− embryos (Figure 6(a)–(d)). However, in contrast to single Nfia−/− or Nfib−/− brains, we observed scattered GFAP-positive cells in the vicinity of the midline, which may represent midline zipper glia or indusium griseum glia (Figure 6(a)–(d)). As their presence and locations varied between individual embryos, these cells are likely to represent individual cells that escaped recombination and therefore maintained NFIB expression as previously described (Harris et al., 2016). In single heterozygous knockout embryos, all three glial populations were present, although GFAP expression levels were reduced (Data not shown; Shu et al., 2003; Steele-Perkins et al., 2005). GFAP expression was also reduced in the ammonic neuroepithelium and fimbrial glioepithelium of the hippocampus of Nfia+/−;Nfib+/− mice. This phenotype was more reminiscent of Nfia−/− embryos than Nfib−/− embryos, in which GFAP expression was retained to a greater extent in the fimbrial glioepithelium (Figure 6(e)–(h)). Hence, the loss of one allele of both Nfia and Nfib to a large extent mimics the glial phenotype of homozygous knockout of either family member.

Complete allelic loss of Nfia and Nfib results in a more severe cortical phenotype
The question remains as to whether a further reduction in the number of Nfia and Nfib alleles would affect corticogenesis. To assess whether loss of both copies of Nfia and Nfib increase the severity of the developmental brain phenotype, we analysed Nfia;Nfib double homozygous knockout embryos at E16. Compared to double heterozygous (Figure 7) or single homozygous knockout embryos (Barry et al., 2008; Das Neves et al., 1999; Piper et al., 2009, 2010; Shu et al., 2003; Steele-Perkins et al., 2005), the phenotype of double homozygous knockout embryos was more severe. In contrast to the other genotypes examined, no GFAP-positive cells were detected at the midline of Nfia−/−;Nfib−/− mice (Figure 7(a′)–(c′)), demonstrating a further reduction in glial differentiation in these mice. In the hippocampus, the fimbria of double homozygous knockout animals showed even more prominent decrease in glial differentiation, with no GFAP-positive cells detected (Figure 7(d)–(f)).

The lateral ventricles of these mice were consistently enlarged compared to the other genotypes (Figure 7(a)–(c)) (Steele-Perkins et al., 2005; Das Neves et al., 1999). In conjunction, the ventricular length of both the cingulate cortex and neocortex increased in double knockout embryos, resulting in a near doubling of length in comparison with the wildtype, single knockout and heterozygous double knockout animals (Figure 7(g) and Table 5). Although the overall thickness of the cortex remained unchanged when compared to single knockout or double heterozygous animals, cortical plate thickness was reduced in the medial neocortex (Figure 7(c)″ and Table 5). The medial neocortex of double knockout embryos therefore consists of relatively more germinal zone when compared to the lateral neocortex (Figure 7(a)″–(c)″ and Table 5). This change in layer contribution to overall cortical thickness is consistent with the high medial to low lateral expression gradient of NFIA and NFIB (Bunt et al., 2015). The more severe phenotype observed in the medial neocortex, quantified as the ratio of the cortical plate versus the germinal zone, is consistent with higher endogenous NFIA and NFIB expression in the medial neocortex as compared to the lateral neocortex (Figure 7(h) and Table 5). Furthermore, the hippocampal germinal zone was enlarged in double homozygous mice (Figure 7(i)).
Quantification of cortical measurements.

Additional staining for the neuronal marker TBR1 revealed that cortical lamination was disrupted within Nfia−/−;Nfib−/− mice, with an apparent absence of the subplate (Figure 8(a)–(c)). We also observed severe perturbations in axon projections within the neocortex (Figure 8(d)–(f)″). No GAP43-positive axons were observed projecting to the midline in Nfia−/−;Nfib−/− mice (Figure 8(f)″) and the white matter within the neocortex was reduced (Figure 8(f)′). The remaining projections observed in the neocortex displayed an abnormal morphology and some ectopically projected to or from the marginal zone. Hence, the lack of both Nfia and Nfib culminates in cortical phenotypes of increased severity compared to those resulting from individual Nfi deletion.

Discussion
This work has revealed that during embryogenesis, NFIA and NFIB function additively to regulate cortical development, as decreasing the number of Nfi alleles culminated in a more severe cortical phenotype. This is in line with the overlap of NFIA and NFIB functions in regulating radial glial proliferation and differentiation (Barry et al., 2008; Betancourt et al., 2014; Gobius et al., 2016; Piper et al., 2009, 2010, 2014), with a further reduction in the production of differentiated neural and glial progeny evident when both genes are knocked out.
Whether NFIA and NFIB proteins can fully recapitulate each other’s biological function in the radial glia during corticogenesis remains to be further investigated. As not all misregulated genes in the Nfia and Nfib knockouts overlap, it is not clear whether only the shared misregulated genes are responsible for the observed phenotypes, or whether NFIA and NFIB also uniquely regulate other downstream targets that contribute to the observed phenotype. The latter is complicated as NFIA and NFIB are differentially expressed in the progeny of radial glia. Therefore, the differences in regulated genes detected in our transcriptomic analyses might originate from differentiated cells, rather than from the radial glia themselves. This could explain why the unique NFIA or NFIB targets were not enriched within the VZ datasets. However, the shared down-regulated genes are enriched for direct NFI binding and high expression in the VZ (Tables 1 and 2). For a subset of the potential direct targets in radial glia, such as Bcan, Ccdc80, Slc26a7 and Slc9a3r, the VZ expression also showed a high caudal to low rostral expression gradient (Allen Institute, 2013; Diez-Roux et al., 2011; Kawaguchi et al., 2008; Visel et al., 2004), similar to NFIA and NFIB (Bunt et al., 2015). Despite this, these potential direct targets might not be sufficient to explain the complete Nfi knockout phenotype. For instance, lectican protein BCAN and extracellular matrix glycoprotein tenascin-C (TNC) are implicated with altered radial glial proliferation and differentiation (Garcion et al., 2004), but combined knockout of these genes together with other family members did not result in a severe developmental brain phenotype (Rauch et al., 2005). Nevertheless, these shared down-regulated genes provide a foundation to investigate the role of NFIA and NFIB in radial glial differentiation. Gene expression analyses using mRNA isolated specifically from the radial glia of Nfia and Nfib knockout embryos, rather than whole neocortex, will be required to fully dissect the regulated pathway and the potential overlap in targets between the two proteins. The exclusion of other cell types of the neocortex, such as intermediate progenitors, neurons and glia, will provide a more complete picture of both common and distinct functions of these genes, especially if combined with chromatin immunoprecipitation studies to determine direct binding.
In vitro experiments have demonstrated that the DNA-binding motif of both proteins are near identical (Jolma et al., 2013), suggesting that NFIA and NFIB likely bind to the same target genes. However, we cannot rule out that their relative binding affinity may differ in vivo. More importantly, the different C-terminal transactivation domains of NFIA and NFIB might vary in how they interact with other proteins and hence how they activate their target genes, thus contributing to the observed differences in our mRNA-sequencing datasets. It is possible that genes that are uniquely misregulated in only one knockout might represent transcriptional targets for which this specific NFI has the strongest DNA binding affinity/specificity or has stronger transcriptional activation. Such genes may still be regulated by the other NFI family member, but due to their lower affinity or activation, the observed regulation may be masked when the other NFI family member is present.
Based on the high similarity of the double heterozygous knockout brains with either single homozygous Nfia or Nfib knockout brains, we can conclude that in early development, both NFIA and NFIB function very similarly in corticogenesis and therefore may, in part, compensate for one another when there is a reduction in copy number or functional mutation in one family member. Our data also demonstrate that no specific dimer is essential for cortical development. Although displaying similar phenotypes, in single homozygous knockout mice only NFIA or NFIB homodimers can exist, but in double heterozygous knockouts all homo- and heterodimers can form. Given that double homozygous mice have a more severe phenotype, the different types of dimers seem to function similarly in cortical development.
However, at a later developmental stage, when neurogenesis and gliogenesis are more pronounced, both NFIA and NFIB or specific dimers might function in distinct roles. In the adult brain, mature astrocytes express both NFIA and NFIB, whereas mature oligodendrocytes mainly express NFIA and neurons express NFIB (Chen et al., 2017). In vitro, both NFIA and NFIB together with SOX9 are required for astrocytic differentiation from induced pluripotent stem cells (Caiazzo et al., 2015). Furthermore, altering the level of NFIA has also been shown to affect astrocytic and oligodendrocytic gene expression (Glasgow et al., 2014; Wong et al., 2007). Thus, changes in their relative expression might play a more significant role in neurons and glia, at later stages of cortical development and in the adult brain.
Our current lack of knowledge about the regulation of Nfia and Nfib expression limits our understanding of their role in cerebral development. Why Nfia and Nfib expression patterns are similar in radial glia, but not in their neuronal and glial progeny, remains unclear. No upstream regulatory transcription factors for Nfi have been identified that would explain their initial expression at E12, although their high caudo-medial to low rostro-lateral expression pattern (Bunt et al., 2015) suggests that they are likely to be positively regulated by transcription factors with a similar expression pattern. Alternatively, post-transcriptional regulation could be responsible for this distinct expression gradient. For example, miR-153 (Tsai et al., 2014; Tsuyama et al., 2015) and Drosha (Rolando et al., 2016) have been shown to regulate Nfi mRNA levels and translation during neurogenesis. How this similar expression pattern in radial glia subsequently transitions to a cell type-specific glial and neuronal expression remains to be identified.
Due to the perinatal lethality of Nfia and Nfib knockout mice, as well as the low embryonic viability of Nfia−/−;Nfib−/− mice used in our study, these models have limited applicability to determine the progression and functional consequences of the observed phenotype postnatally. Furthermore, these models cannot answer fundamental questions about the differences in function of NFIA and NFIB or their dimers. Brain-specific deletion of both Nfi genes using conditional knockout alleles may overcome some of these issues, assuming that double homozygous knockout in the brain has no effect on viability. In theory, utilising the recent generation of a conditional Nfib overexpression model (Semenova et al., 2016) crossed to an Nfia knockout mouse would be able to demonstrate whether increasing NFIB protein levels could compensate for the absence of Nfia. However, this would require ectopic NFIB expression to mimic both the expression level and temporal and spatial specificity of NFIA. A better approach would be a knock in of Nfia into the Nfib locus or vice versa, although this might cause problems in other tissues where a specific family member might be required.
The question remains whether NFIA and NFIB have only overlapping functions in regulating radial glial proliferation and differentiation or whether they also have yet unknown distinct roles in this cell type. If they function interchangeably in radial glia, their combined expression might provide a more precise mechanism to regulate the balance between proliferation and differentiation. As we show that loss of NFI results in a dosage-dependent cortical defect, maintaining the correct protein levels is essential for normal cortical development. In conclusion, our work demonstrates that NFIA and NFIB function additively in the context of early cerebral development, as the combined allelic loss of these genes directly correlates with the severity of the developmental brain phenotypes.
Footnotes
Acknowledgements
The authors thank the staff of the University of Queensland Biological Resources (UQBR) animal facility, the QBI Advanced Microscopy and Analysis Facility and the QBI Centre for Brain Genomics for their expertise and assistance in this project. The authors are grateful to Rowan Tweedale for her critical comments on the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
This work was supported by National Health and Medical Research Council project grants (GNT1057751 to M.P. and GNT1100443 to L.J.R.), Australian Research Council Discovery Grants (DP160100368 to M.P. and DP140101499 to L.J.R.) and NYSTEM grants (C026429 and C030133 to R.M.G.). M.P. was supported by a fellowship (Australian Research Council Future Fellowship; FT120100170). L.J.R. was supported by an NHMRC Principal Research Fellowship. D.V., J.W.C.L. and L.H. were supported by an Australian Government Research Training Program Scholarship. J.W.C.L. was supported by an UQ Centennial Scholarship. Y.Y. was supported by the Aleks Brumby Summer Research Scholarship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders.