
Abstract
Background:
En coup de sabre (ECDS) is a rare variant of localized scleroderma which can be associated with neurologic symptoms including seizures, focal neurologic deficit, and movement disorders. Little is known about the disease course in ECDS. We describe two patients with a history of localized scleroderma ECDS of the scalp who developed worsening neurologic symptoms coincidental in timing with extension of preexisting skin and skull lesions and evidence of inflammatory pathology on brain biopsy.
Case Report:
Our first patient was diagnosed with localized scleroderma ECDS at the age of 12. He re-presented at age 34 with progression of his skin lesions, new indentation of the occipital bones, and new neurologic symptoms of ataxia, cranial nerve IV palsy, and cognitive decline. MRI brain revealed multiple lesions that were hyperintense on T2-weighted images, the largest of which was in the left temporal lobe. Brain biopsy pathology was consistent with a lymphocytic inflammatory process. He was treated with pulse steroids and mycophenolate with stabilization of his symptoms and brain lesions on imaging. Our second patient was initially diagnosed with localized scleroderma ECDS at age 46. Concurrently, neuroimaging showed right frontal and temporal lesions that were hyperintense on T2-weighted images in the context of neurologic symptoms of vertigo and headache. At age 50, he developed worsening of existing skin lesions followed by new generalized tonic-clonic seizures and behavioral changes. MRI brain revealed worsening of his brain lesions, and ultimately brain biopsy confirmed focal perivascular lymphocytic reaction consistent with immune-mediated vasculitis. He was treated with pulse steroids and cyclophosphamide with improvement in his symptoms.
Conclusion:
Neurologic symptoms associated with localized scleroderma are a rare but well-documented association. These cases highlight the need to consider diffuse intracranial inflammatory pathology, rather than simply localized brain lesions, in ECDS, particularly in instances where skin lesions are progressive.
Keywords
Introduction
Localized scleroderma, or morphea, is an inflammatory condition that encompasses a wide variety of clinical phenotypes. Skin lesions are characterized by areas of skin thickening associated with induration, hyperpigmentation, and increased fibrous tissue. Localized scleroderma can be subdivided into five types: circumscribed morphea, linear scleroderma, generalized morphea, pansclerotic morphea, and mixed morphea. 1 A further subdivision can be made for linear scleroderma into disease involving the trunk/limbs, and en coup de sabre (ECDS), which affects the head. 1
The estimated incidence of localized scleroderma in adults is 3.25/100,000 person years. 2 ECDS is a variant with typical skin lesions affecting the paramedian forehead. ECDS is more commonly reported in the pediatric population with a prevalence of 3%–17.6% in localized scleroderma studies compared to 2.4%–4.4% in adult studies. 3 In both adults and pediatrics, it has been associated with a variety of neurologic symptoms including headaches, seizures, focal neurologic deficit, ophthalmologic abnormalities, and movement disorders.4,5 Compared to the adult population, pediatric patients are also more likely to have extracutaneous manifestations with an estimated incidence of 4% and a less favorable disease course.1,3,4
Little is known about the disease course, preferred treatment, and pathophysiology of neurologic involvement in adults with ECDS given the scarce number of reported cases with histopathologic correlation on brain biopsy in addition to neuroimaging.
In our case study, we describe two patients with a history of localized scleroderma ECDS who developed worsening neurologic symptoms after deepening of their preexisting skin lesions. In both cases, there was evidence of multifocal intraparenchymal brain involvement on imaging, inflammatory pathology was identified on brain biopsy, and they were treated with systemic immunosuppression. One of our cases also had bilateral skin and brain lesions. These cases demonstrate the variability of intracranial brain lesions in ECDS and suggest that clinicians should be aware of progressive or deepening lesions, as it may indicate impending worsening of neurologic symptoms.
Case 1
A 37-year-old man presented to an outpatient rheumatology clinic with unexplained neurologic symptoms and brain lesions in the context of progressive linear scleroderma with new lesions to the occiput. He was initially diagnosed with linear scleroderma involving the forehead at the age of 12 and was treated with D-penicillamine for an unknown period of time. Six months prior to being seen in our clinic, he was also started on prednisone (1 mg/kg) with taper and maintained on prednisone 10 mg daily for query multiple sclerosis as the etiology for his neurologic symptoms. His history was significant for a 2-year history of ataxia, cranial nerve IV palsy, and severe, progressive, cognitive decline including memory loss, bowel incontinence, anxiety, and the loss of ability to perform activities of daily living. His family described deepening of his forehead lesions as well as a new lesion to his occiput prior to the onset of these neurologic symptoms. Differential diagnoses at the time included tumefactive multiple sclerosis (although he was previously treated with Tecfidera with no benefit), Parry-Romberg syndrome, viral or autoimmune encephalitis, and neurosarcoidosis. He was otherwise systemically well without symptoms of systemic sclerosis or other connective tissue disorder. His physical exam revealed U-shaped linear scleroderma over the scalp with a 10-cm patch over the left scalp and an 8-cm patch over the right scalp area as well as an area of linear scleroderma in the occiput (Figure 1). The rest of his skin examination revealed no sclerodactyly, typical telangiectasias, or nailfold changes. Cognitive testing revealed reduced executive functioning, working memory, self-awareness, and insight and noted impulsivity. His initial rheumatologic workup revealed negative antinuclear antibodies (ANA), anti neutrophil cytoplasmic antibodies (ANCA), rheumatoid factor, and anti-myelin-associated glycoproteins and normal complements, c-reactive protein (CRP), and angiotensin converting enzyme (ACE) levels. Chest x-ray and computed tomography (CT) of the chest did not reveal any evidence of granulomatous disease. Autoantibody panels for paraneoplastic antibodies and autoimmune encephalitis were negative. He had multiple cerebrospinal fluid analyses throughout his course, the initial showing non-matching oligoclonal banding and elevated protein at 0.7 g/L, but with subsequent CSF results not replicating this finding. His MRI brain at the onset of symptoms showed multiple hyperintense subcortical lesions within the left temporal and frontal lobe on axial T2-weighted and fluid-attenuated inversion recovery (FLAIR) images and punctate foci of enhancement on post-gadolinium T1-weighted images (Figure 2). A brain biopsy was taken of the temporal lobe lesion, with findings of focal areas of increased perivascular CD3+ and CD8+ T-cell lymphocytes within the tissue (Figure 2). Over the years, there has been periodic improvement in some lesions, as well as new lesions on serial imaging.
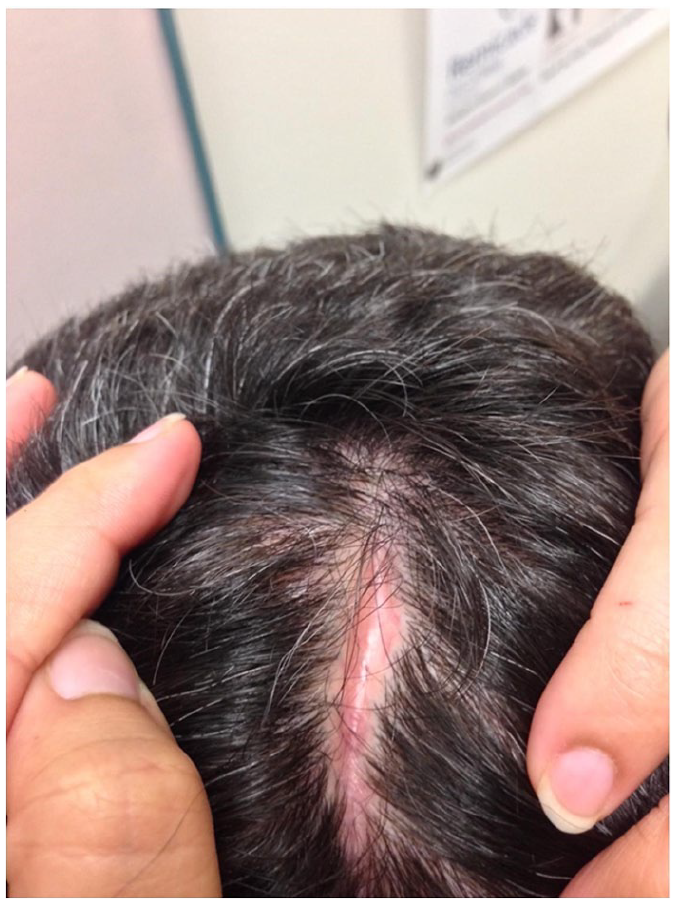
Localized scleroderma of the occiput.
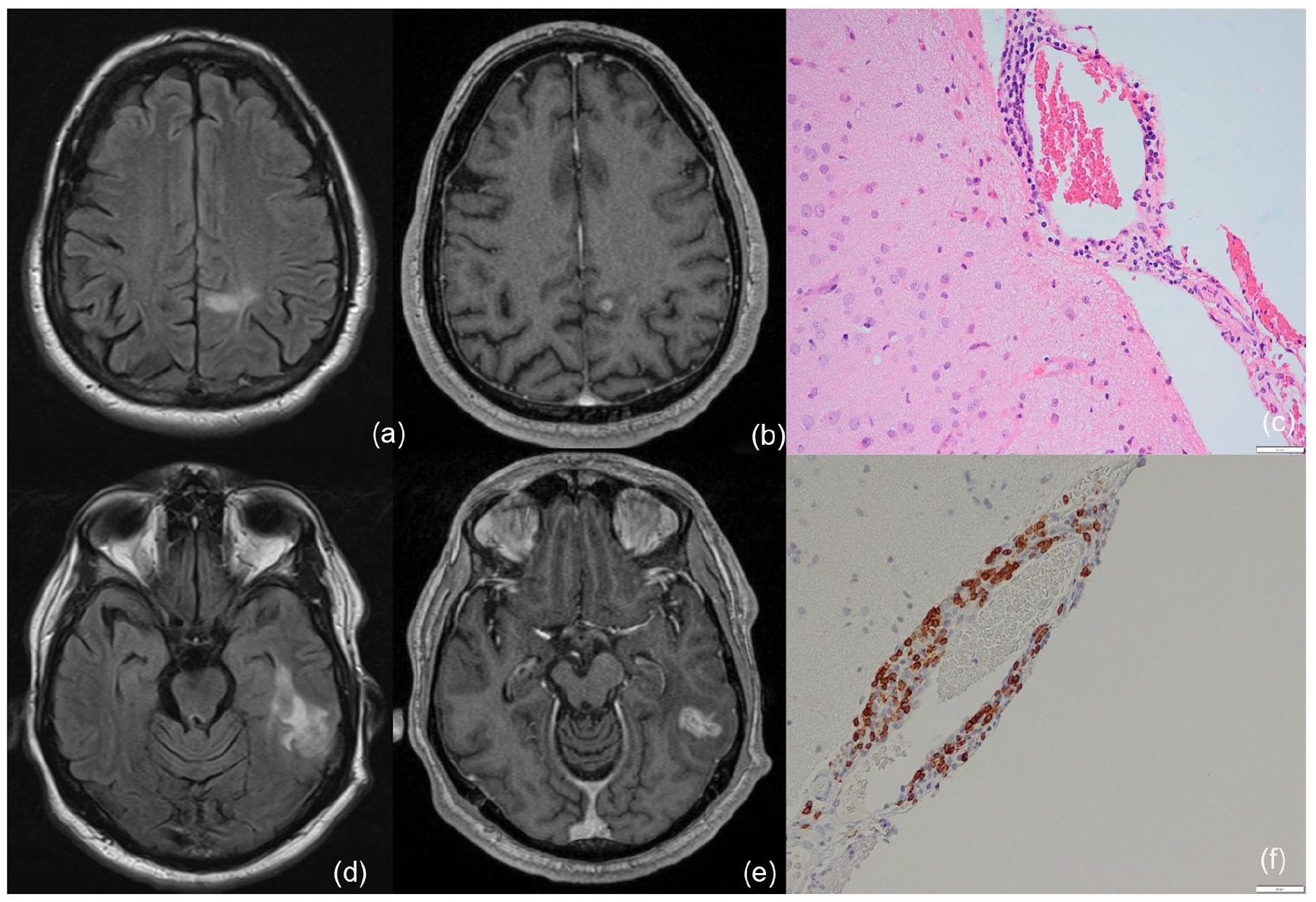
At initial presentation, FLAIR images exhibit a small irregular hyperintense subcortical lesion within the left frontal lobe (a) and a large irregular hyperintense subcortical lesion within the left temporal lobe (d). The left frontal lesion contains a punctate focus of enhancement on post-gadolinium T1-weight images (b), while the left temporal lesion contains a small central ovoid focus of enhancement on post-gadolinium T1-weighted images (e). Brain biopsy pathology exhibit meningeal lymphocytic inflammation with prominent perivascular focus (c) and lymphocytic inflammation consisting largely of CD3+ T cells (f).
The patient was initially started on methotrexate (20 mg PO weekly), but this was discontinued due to elevated liver enzymes. He was maintained on prednisone 10 mg PO daily with slow taper. He was briefly treated with oral cyclophosphamide (100 mg daily), which was discontinued for a perception of lack of improvement in symptoms as well as a question of viral infection as the cause for his brain lesions, which was ultimately ruled out in conjunction with local infectious disease consultants.
One year later, he developed worsening progressive neurologic decline as well as generalized tonic-clonic seizures. Repeat MRI brain demonstrated new subcortical lesions involving the left frontoparietal region and the bilateral temporal lobes. Immunosuppression was re-initiated with mycophenolate mofetil (1500 mg twice daily) and oral prednisone (5–10 mg daily).
Since initiating mycophenolate, both the patient’s neuropsychiatric symptoms and skin lesions have remained stable over a 6-year period. He has surveillance MRIs every year, which have not shown any new lesions.
Case 2
A 46-year-old man presented with two linear hyperpigmented skin lesions of the right forehead consistent with morphea/ECDS lesions. He had no relevant personal or family history. He did not have any clinical features suggestive of systemic sclerosis or other connective tissue disorder. His physical exam was unremarkable aside from the linear scleroderma over the forehead.
Concurrently, the patient described recurrent episodes of vertigo, syncope, and severe migraine headaches, for which he was referred to an outpatient neurology clinic. An MRI brain with gadolinium-enhanced sequences demonstrated multifocal right-sided subcortical white matter FLAIR-hyperintense lesions, the largest of which was in the right frontal lobe with tiny foci of enhancement. Differential diagnosis at the time of presentation included a neoplastic process such as lymphoma, vasculitis, or an infectious process such as tuberculosis. Extensive workup for these brain lesions did not reveal an alternate diagnosis. Lumbar puncture was completed, and the cerebrospinal fluid glucose, protein, and white blood cell levels were within normal limits. The CSF was negative for bacterial culture, viral culture, malignant cells, paraneoplastic antibodies, and oligoclonal bands. His rheumatological serologies were normal, including ANCA testing, ANA panel by multiplex immunoassay, rheumatoid factor, complement levels, and CRP. Chest x-ray and CT of the chest did not reveal any evidence of granulomatous disease. He was initially treated with hydroxychloroquine (400 mg daily), topical corticosteroids, and ultraviolet phototherapy by his dermatologist. For further diagnostic clarification, a brain biopsy was recommended to the patient. However, this was deferred for patient preference. In the interim, he had serial imaging of his brain with MRI showing spontaneous improvement in some brain lesions as well as some improvement in his headache symptoms.
Three years after his initial presentation, he developed generalized tonic-clonic seizures and frank encephalopathy requiring admission to hospital. Six months prior, he had been seen in clinic and noted to have deepening of his preexisting skin lesions, with marked skull depression on the forehead on physical exam. Repeat CSF analysis demonstrated slightly elevated glucose, at 4.1 mmol/L, and white blood cell count of 17, but, otherwise, protein was within normal limits, and fluid was negative for malignant cells, oligoclonal bands, bacterial, viral, and tuberculosis culture. Repeat brain MRI with gadolinium-enhanced sequences showed interval enlargement of a lesion in the right temporal lobe with focal enhancement and vasogenic edema (Figure 3(d)). A brain biopsy was expedited and showed significant perivascular lymphocytic reaction with transmural involvement and a few scattered perivascular foci and scattered reactive T cells present, which was felt to be consistent with immune-mediated vasculitis (Figure 3(f)). He was treated with 3 days of IV pulsed steroids (1 g daily) followed by oral prednisone at a dose of 1 mg/kg with a slow taper. He was then induced with IV cyclophosphamide following the CYCLOPS protocol, 6 followed by mycophenolate mofetil (1 g twice daily) as a maintenance agent in addition to low-dose prednisone (5 mg daily). Since starting immunosuppression, his symptoms have improved, and the vasogenic edema and enhancement seen on surveillance MRI has also improved with no new lesions or interval progression in previous lesions.
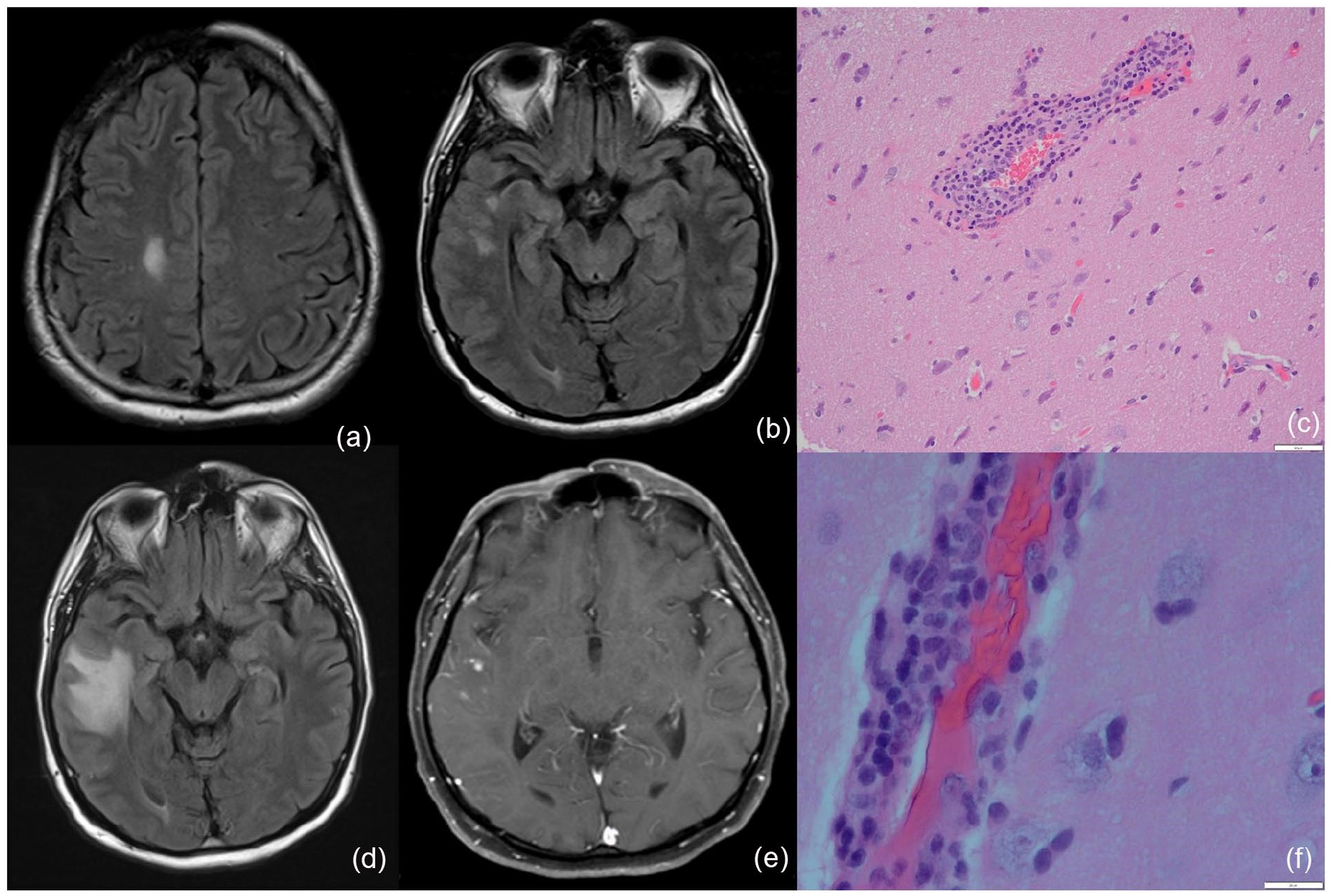
At initial presentation, FLAIR images show a moderately-sized region of scalp and calvarial thinning within the right frontal bone (a), a small hyperintense subcortical lesion within the right frontal lobe (a) and two small hyperintense subcortical lesions within the right temporal lobe (b). Three years after the initial presentation, on axial FLAIR images, the right temporal lobe lesions have increased in size, forming one large cortical lesion (d), which also contains several punctate foci of enhancement on post-gadolinium-T1-weighted images (e). The right temporal lesion is also associated with mild volumetric expansion, indicative of vasogenic edema. The degree of scalp and calvarial thinning is unchanged (not shown). Brain biopsy pathology exhibits marked perivascular mononuclear/lymphocytic inflammation (c, f) with some transmural involvement (f).
Conclusions
Neurologic symptoms are a well-documented association with scleroderma ECDS that rheumatologists and neurologists should be aware of. However, previously published case reports have more commonly demonstrated an association with intraparenchymal white matter lesions that are solitary and ipsilateral to the skin lesion. Our case highlights the less-common association with multifocal and contralateral brain lesions. 7 Documented neuropathology of these lesions are scarce, and a variety of pathologies have been demonstrated with both inflammatory changes, including perivascular lymphocytic infiltrate as well as cases of non-inflammatory changes with leptomeningeal band-like sclerosis and ectatic vessels.8–10 In our cases, both biopsies demonstrated a significant mononuclear inflammatory reaction involving the meninges and underlying brain parenchyma with areas of perivascular accentuation. Focally, this led to transmural vessel involvement (i.e. vasculitis), although without fibrinoid necrosis in the areas sampled. The inflammatory reaction was predominantly T lymphocytes (CD3) enriched in the cytotoxic T-cell subset (CD8) with an associated macrophage reaction without B-cell involvement. This process was non-granulomatous, distinct from neurosarcoidosis, and suggests an immune/autoimmune process variably affecting the meninges and brain parenchyma.
Our case reports add to the current literature surrounding scleroderma ECDS with neurologic involvement by highlighting two features: (1) skin and brain lesions can be bilateral and multifocal and demonstrate neuropathologic evidence of immune-mediated vasculitis; and (2) deepening of skin lesions preceded worsening neurologic involvement and should be considered as a risk factor for inflammatory central nervous system involvement.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.