
Abstract
Systemic sclerosis (SSc) is a chronic autoimmune disorder characterized by progressive fibrosis, vasculopathy, and immune dysregulation. The disease commonly presents with Raynaud’s phenomenon and skin thickening, commonly of the upper limb. Isolated lower extremity presentation is uncommon and often misdiagnosed. This diagnostic uncertainty may lead to delayed recognition and increased morbidity, particularly when SSc mimics lymphedema in the early stages. We report a case of a 61-year-old female who initially presented with lower extremity swelling and was misdiagnosed with primary lymphedema. Despite treatment with diuretics and compression therapy, her symptoms progressed to involve her upper extremities, prompting further evaluation. Physical examination revealed non-pitting lower extremity scleroderma, sclerodactyly, puffy hands, and nailfold capillary abnormalities. Laboratory workup was positive for anti-RNA polymerase III antibodies, a marker associated with an increased risk of scleroderma renal crisis. The patient developed scleroderma renal crisis after a delayed diagnosis, necessitating hospital admission and initiation of angiotensin-converting enzyme inhibitors. This case highlights the challenges in distinguishing early lower extremity SSc from lymphedema. Early identification of atypical SSc presentations is critical to be cognizant of life-threatening complications such as scleroderma renal crisis. Clinicians should maintain a high index of suspicion for SSc in patients with persistent non-pitting lower extremity swelling, skin thickening, and abnormal capillaroscopy findings, even in the absence of initial upper limb involvement.
Keywords
Introduction
Systemic sclerosis (SSc), commonly known as scleroderma, is a progressive autoimmune disorder characterized by fibrosis, microvascular dysfunction, and immune dysregulation. While SSc typically manifests with Raynaud’s phenomenon and upper extremity skin involvement, lower extremity onset is uncommon and can lead to diagnostic confusion, particularly with conditions such as lymphedema. 1
Lymphedema is a distinct disorder caused by impaired lymphatic drainage, with early pitting edema but leading to progressive non-pitting edema, fibrosis, and tissue hypertrophy. 2 Unlike SSc, it is not an autoimmune condition and lacks microvascular compromise or systemic organ involvement. However, both conditions share overlapping clinical features, including skin thickening, swelling, and fibrosis, which can make differentiation challenging, particularly in the early stages. 1
Recent studies have documented lymphatic vessel loss in scleroderma, particularly in affected skin regions, which may further complicate its distinction from lymphedema. 3 The presence of microvascular abnormalities, such as capillary dropout and digital ulcerations, is a key diagnostic clue that supports SSc rather than lymphedema. 1
Here, we present a case of a patient with atypical lower extremity onset of scleroderma, initially diagnosed as lymphedema and subsequently presenting with scleroderma renal crisis (SRC). This case highlights the importance of recognizing unusual presentations of SSc and the need for careful differentiation between fibrotic skin disorders, particularly when assessing lower extremity involvement.
Case
A 61-year-old female presented to our clinic in May 2024 with a diagnosis of lower extremity lymphedema, having had swelling of legs and feet since November 2023. Presumed lymphedema was being treated with compression stockings and diuretics. She was seen by a rheumatologist, in April 2024, for a positive ANA 1:320, who was suspicious for scleroderma after noting new onset puffiness of the patient’s hands and was referred to dermatology for evaluation of possible scleroderma. A fortnight before presenting to the clinic, the patient was hospitalized because of gastrointestinal (GI) bleeding and required a blood transfusion. From which the patient developed a transfusion reaction and required oral glucocorticoids. Outpatient endoscopy, after discharge, showed gastric erosions. However, no concern was raised for gastric antral vascular ectasia as the underlying cause of the patient’s GI bleeding. Since being discharged from the outside hospital, the patient reported having higher blood pressure than usual at home, with systolic reported as in the 170s to 180s. The patient was seen in the rheumatology-dermatology clinic 10 days after discharge from the outside hospital.
At the initial rheumatology-dermatology clinic visit, the patient’s blood pressure was 180/85 mm Hg. On physical exam, the patient was found to have puffy hands, feet, and legs with sclerodactyly and scleroderma (Figure 1). Nailfold capillaroscopy revealed dilatation and looping of capillaries, as well as dropout of capillaries. Of note, the patient did not complain of Raynaud’s symptoms.
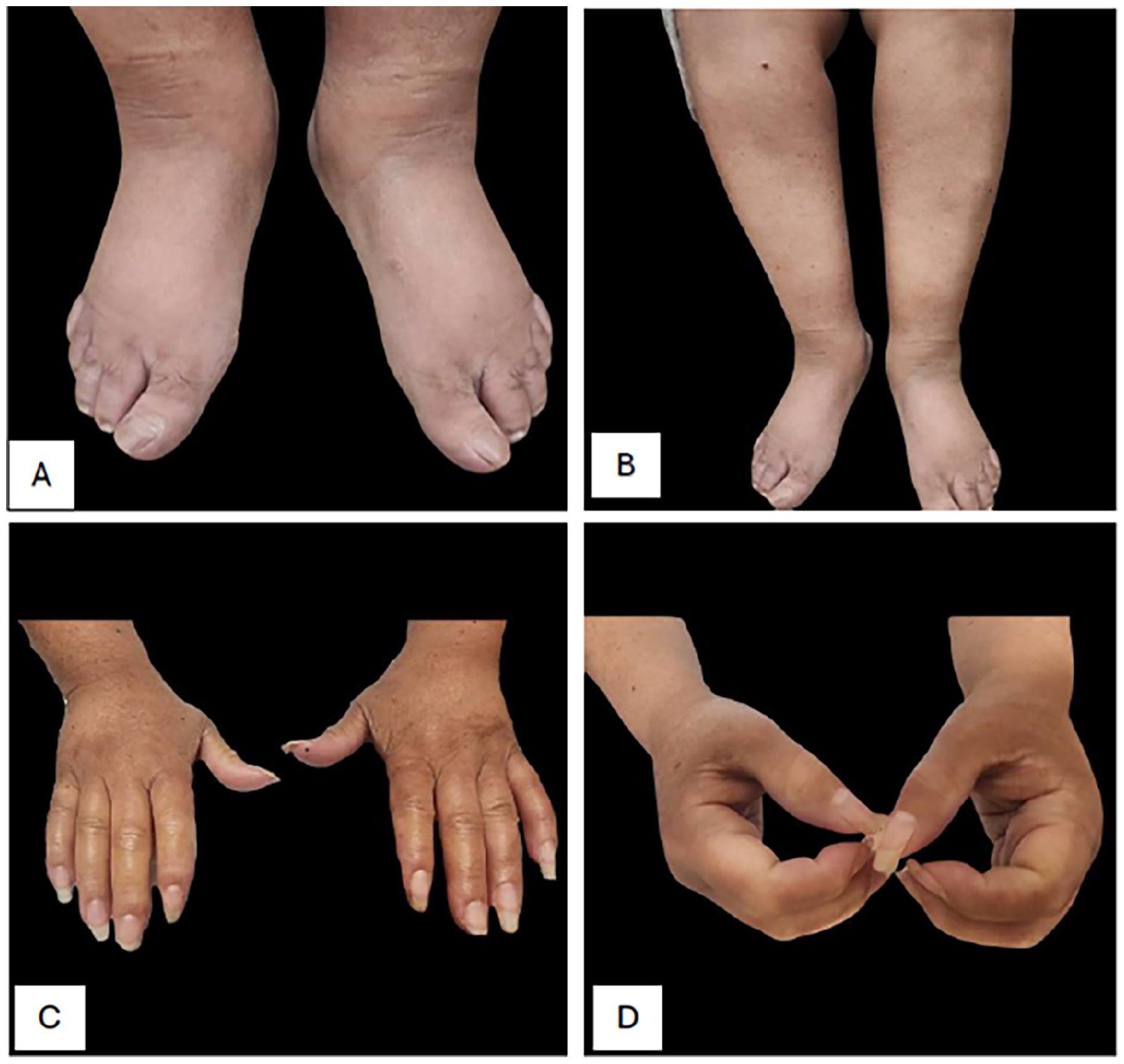
(A) Scleroderma affecting the dorsum of bilateral feet. (B) Scleroderma involving bilateral lower legs. (C and D) Puffiness of bilateral hands, sclerodactyly distal to PIP joints.
The patient’s creatinine jumped from 0.6 to 1.87 mg/dL (0.44–1.03 mg/dL) in 1 week since discharge from the hospital. The patient was being diuresed for presumed lymphedema with bumetanide. Given the constellation of symptoms, blood work abnormalities and hypertension post-steroid use along with precipitous worsening of kidney function, a diagnosis of SSc with SRC was made, and the patient was admitted to the hospital for immediate management with short-acting angiotensin-converting enzyme (ACE) inhibitors. The patient’s RNA polymerase III test returned positive (>80 U; normal < 20 U), which carries with it an increased risk of SRC. The patient was negative for SSA, SSB, smith, smith/RNP, RNP, dsDNA, ANCA, and normal complement levels. The patient’s blood pressure was well controlled on an ACE inhibitor and was begun on treatment with mycophenolate mofetil which was up-titrated to 3 g/day. Pulmonary function tests, computed tomography (CT) chest, and echocardiogram of the heart, as part of the SSc workup, did not reveal any abnormalities.
Discussion
La Montagna et al. 4 studied the prevalence of skin involvement of upper and lower extremities in SSc and found that skin thickening is detected less frequently in feet than in hands (26% vs 96%; p < 0.001) and with lower partial skin thickness scores. This was also true when reviewing the literature and online images of scleroderma and edema seen in SSc; examples of pictures are primarily that of fingers and hands and rarely that of the feet and legs.
SSc-related lower extremity involvement is often under-recognized and can resemble chronic lymphedema, particularly in the early edematous phase. 4 However, as the disease progresses, fibrosis, skin tightening, and pigmentary changes become evident, distinguishing it from lymphedema (Table 1). 3
Key diagnostic distinctions between SSc and lymphedema.

Failure to differentiate early lower extremity SSc from lymphedema can lead to significant morbidity. Compression therapy, a mainstay for lymphedema, may exacerbate ischemic damage in SSc patients. 4 Moreover, delayed diagnosis of SSc postpones immunosuppressive treatment, increasing the likelihood of progressive systemic involvement such as interstitial lung disease, pulmonary hypertension, and renal crisis. 5 Given the underlying pathology in SRC is vasculopathy resulting in decreased renal perfusion, treating with ACE inhibitors has been shown to be lifesaving treatment if initiated promptly. 5 Diuretics can potentially worsen hypertension via effects on the renin-angiotensin pathway and therefore are generally considered third-line agents for treatment of SRC behind calcium channel blockers. 5
Conclusion
This case underscores the importance of making a prompt diagnosis. Clinicians should be aware of the overlapping features between lymphedema and early scleroderma. Involvement of feet and legs with skin changes in SSc is important, as the adage goes “the eyes cannot see what the mind doesn’t know”. A multimodal diagnostic approach, integrating clinical assessment, imaging, histopathology, and autoimmune serologies, is critical for early and accurate diagnosis.
Footnotes
Acknowledgements
We would like to thank our patient for the permission to publish this case and our patients in general, whom we learn from on a daily basis.
Correction (June 2025):
The article has been updated with textual changes since the article was published in the issue.
Author contributions
H.A.: Conceptualization, writing, and revising the manuscript.
L.L.A.G.: Writing and revising the manuscript.
T.K.-H.: Writing and revising the manuscript.
A.O.: Revising and editing the manuscript.
X.Y.: Revising and editing the manuscript.
S.C.: Revising and editing the manuscript.
V.P.W.: Supervision and editing the manuscript.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: V.P.W. has grants from Celgene, Janssen, Pfizer, Biogen, Gilead, Corbus Pharmaceuticals, Genentech, AstraZeneca, Viela, Syntimmune, Amgen, Regeneron, Argenx, CSL Behring, Ventus, q32 Bio, BMS, and Horizon and has consulted for Celgene, Genentech, Janssen, Lilly, Pfizer, Biogen, BMS, Gilead, Amgen, Medscape, Nektar, Incyte, EMD Sorona, CSL Behring, Principia, Crisalis, Viela Bio, Argenx, Kyowa Kirin, Regeneron, Principia, AstraZeneca, Abbvie, Octapharma, GSK, AstraZeneca, Cugene, UCB, Corcept, Beacon Bioscience, Rome Pharmaceuticals, Horizon, Gilead, Merck, Kezar, Sanofi, Bayer, Akari, Calyx, and Cabaletta Bio. The University of Pennsylvania owns the copyright for the CLASI. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This manuscript was funded by the National Institutes of Health-USA (NIH-USA) grants R01AR071653 (to V.P.W.) and the United States Department of Veterans Affairs Merit Review BX005921-01 (Veterans Health Administration, Office of Research and Development and Biomedical Laboratory Research and Development, to V.P.W.).
Ethical approval
Not needed; patient consent was obtained.