
Abstract
Objective:
Safe, effective therapies are urgently needed for patients with systemic sclerosis. However, clinical trial recruitment is challenging given the limited number of people with systemic sclerosis and further restrictions imposed by eligibility criteria. Innovative approaches are needed to accelerate development of new therapies. This article describes the rationale and trial design for CONQUEST (NCT06195072), a novel platform clinical trial sponsored by the Scleroderma Research Foundation, a not-for-profit organization.
Methods:
CONQUEST is a multicentre, double-blind, randomized, placebo-controlled, Phase 2b platform trial evaluating the efficacy, safety and pharmacodynamics of multiple investigational products to treat early active systemic sclerosis with interstitial lung disease versus placebo. The primary objective is to evaluate change from baseline to Week 52 in forced vital capacity (mL). Secondary objectives include evaluating changes from baseline to Week 52 in high-resolution computed-tomography-assessed lung involvement and dyspnoea, and overall treatment response (measured using the revised composite response index in diffuse systemic sclerosis score in participants with diffuse cutaneous systemic sclerosis).
Results:
Patients will be enrolled across more than 150 centres in over 25 countries. Recruitment started on 15 April 2024.
Conclusion:
As the first platform clinical trial in the rheumatology field, CONQUEST aims to meaningfully accelerate the development of new therapies for early active systemic sclerosis. Depending on regimen-specific results, trial data could be used to plan and design a Phase 3 trial or may be used alone or together with another registrational trial to establish substantial evidence of effectiveness and safety. The first molecules to be studied, amlitelimab and nerandomilast, both have a strong scientific rationale to modify underlying disease processes in systemic sclerosis.
ClinicalTrials.gov:
Platform Clinical Study for Conquering Scleroderma (CONQUEST); NCT06195072; https://www.clinicaltrials.gov/study/NCT06195072
Keywords
Introduction
Systemic sclerosis (SSc) is a rare, heterogeneous disease characterized by small vessel vasculopathy, production of autoantibodies, fibroblast dysfunction (leading to increased deposition of extracellular matrix), and activation of proinflammatory and profibrotic pathways.1,2 The disease is associated with substantial morbidity, reduced survival and poor quality of life.3–6 Interstitial lung disease (ILD), characterized by inflammation and progressive lung fibrosis, occurs in up to half of patients with SSc early in the course of the disease and is the leading cause of disease-related death.2,7–10
There is a considerable unmet need for safe and effective therapies for SSc, making this an important area for research. 11 However, clinical studies in such a population are complex, time-consuming and resource-intensive, and recruitment is challenging given the limited number of people with SSc who fulfil study enrolment criteria. Innovative clinical studies are needed to accelerate the development of new therapies for people with the condition.
This article describes the rationale and trial design for the CONQUEST trial (ClinicalTrials.gov NCT06195072), which is being sponsored and conducted by the Scleroderma Research Foundation, a not-for-profit organization. This novel Phase 2b clinical platform trial aims to meaningfully accelerate the development of therapy for early active SSc with evidence of ILD. Depending on the data generated in this randomized, placebo-controlled, global setting, trial data could be used to plan and design a Phase 3 trial or may be used alone or with another registrational trial to support the establishment of substantial evidence of effectiveness and safety.
The benefits of the platform clinical trial approach are summarized in Figure 1. Key advantages include the presence of a shared placebo group, which reduces the number of trial participants required in regimen-specific subprotocols, making it ideal for studying rare disease populations. Importantly, to preserve the integrity of randomization, participants will be consented to all regimens available at that time for which they would be eligible, and the intent-to-treat (ITT) analysis data set will include those randomized to receive that regimen as well as only those placebo-control participants who could have been assigned to that specific regimen when they were randomized. The Scleroderma Research Foundation retains ownership of the placebo data, which will be available as a community resource for understanding disease progression, both broadly and within patient subsets. Other efficiencies of the platform trial design include the impact of testing multiple regimens in Phase 2b using shared infrastructure, efficient start-up and common study sites across sub-studies. In addition, the perpetual nature of the platform trial design preserves the study infrastructure for future drugs to be tested.
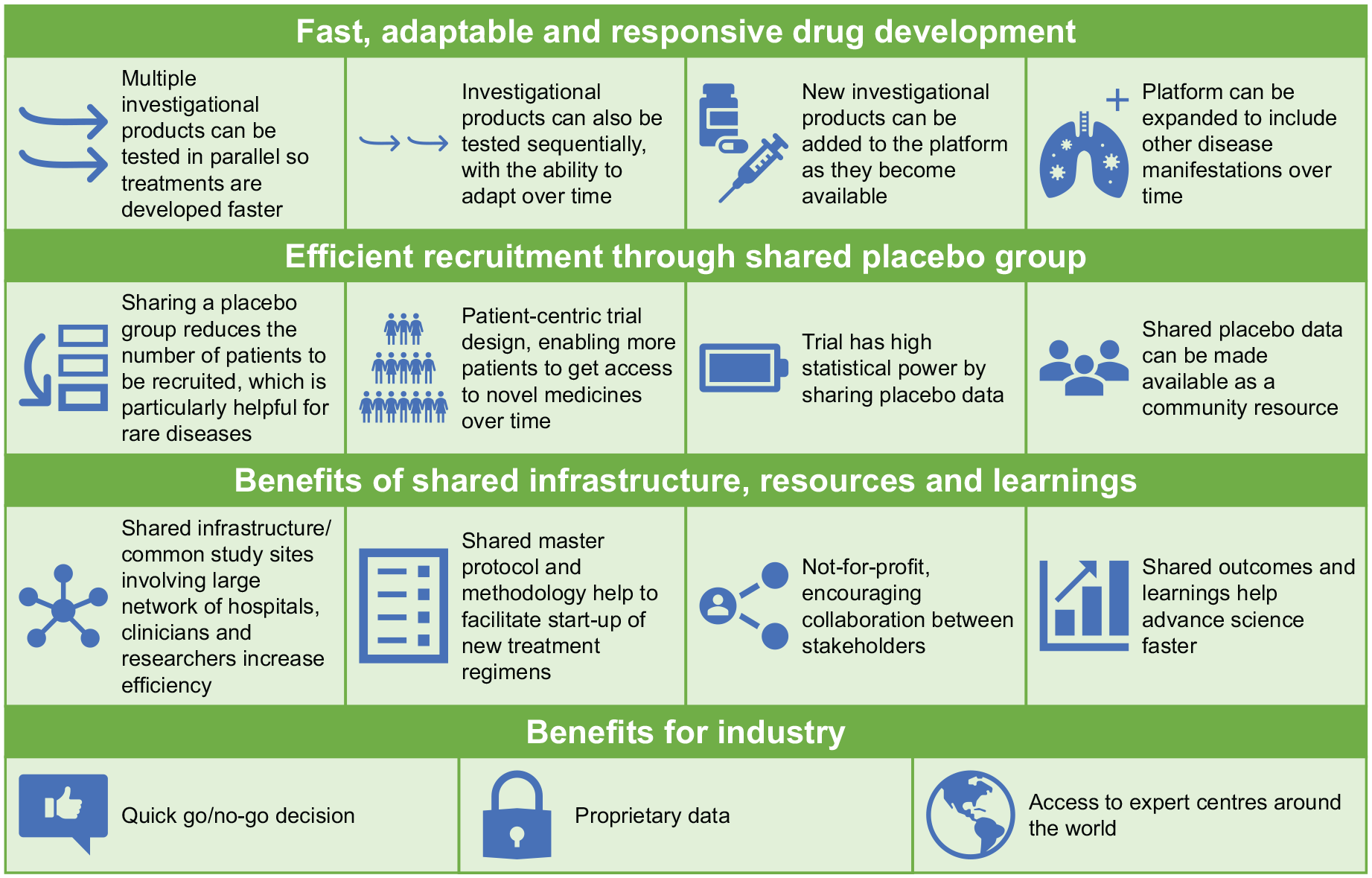
Benefits of the platform clinical trial approach.
CONQUEST is the first platform clinical trial to be carried out in the rheumatology field and is designed to study multiple investigational products (IPs) in parallel and sequentially, with the capability to adapt over time. The platform remains open in the long term and can therefore evaluate new IPs as they become available and allows for the evolution of appropriate endpoints to measure the effect of therapies as they progress.
The first molecules to be investigated in CONQUEST will be amlitelimab, a fully human, non-depleting, non-cytotoxic anti-OX40 ligand monoclonal antibody,12,13 administered by subcutaneous injection, and nerandomilast (BI 1015550), an orally administered preferential phosphodiesterase 4B inhibitor.16,23 Amlitelimab and nerandomilast have had promising results in Phase 2 trials of atopic dermatitis 30 and idiopathic pulmonary fibrosis (IPF), 31 respectively, and nerandomilast is currently being investigated as a treatment for IPF and progressive pulmonary fibrosis in Phase 3 trials.32,33 Both have a strong scientific rationale to modify underlying disease processes in SSc,12–16,18–26,28,29,34–36 as described in Figures 2 to 4.
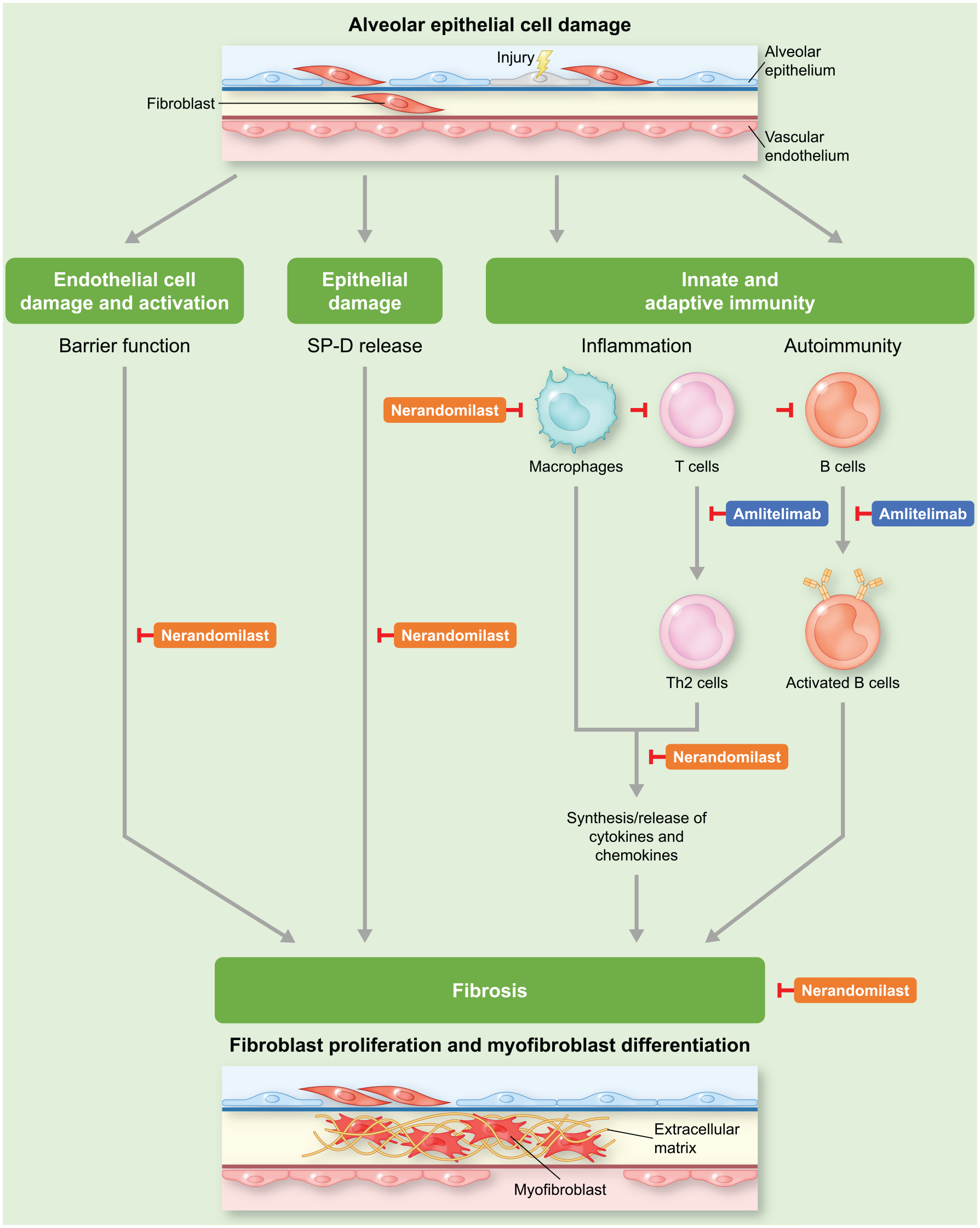
Potential mechanisms of action of amlitelimab and nerandomilast in SSc-ILD.
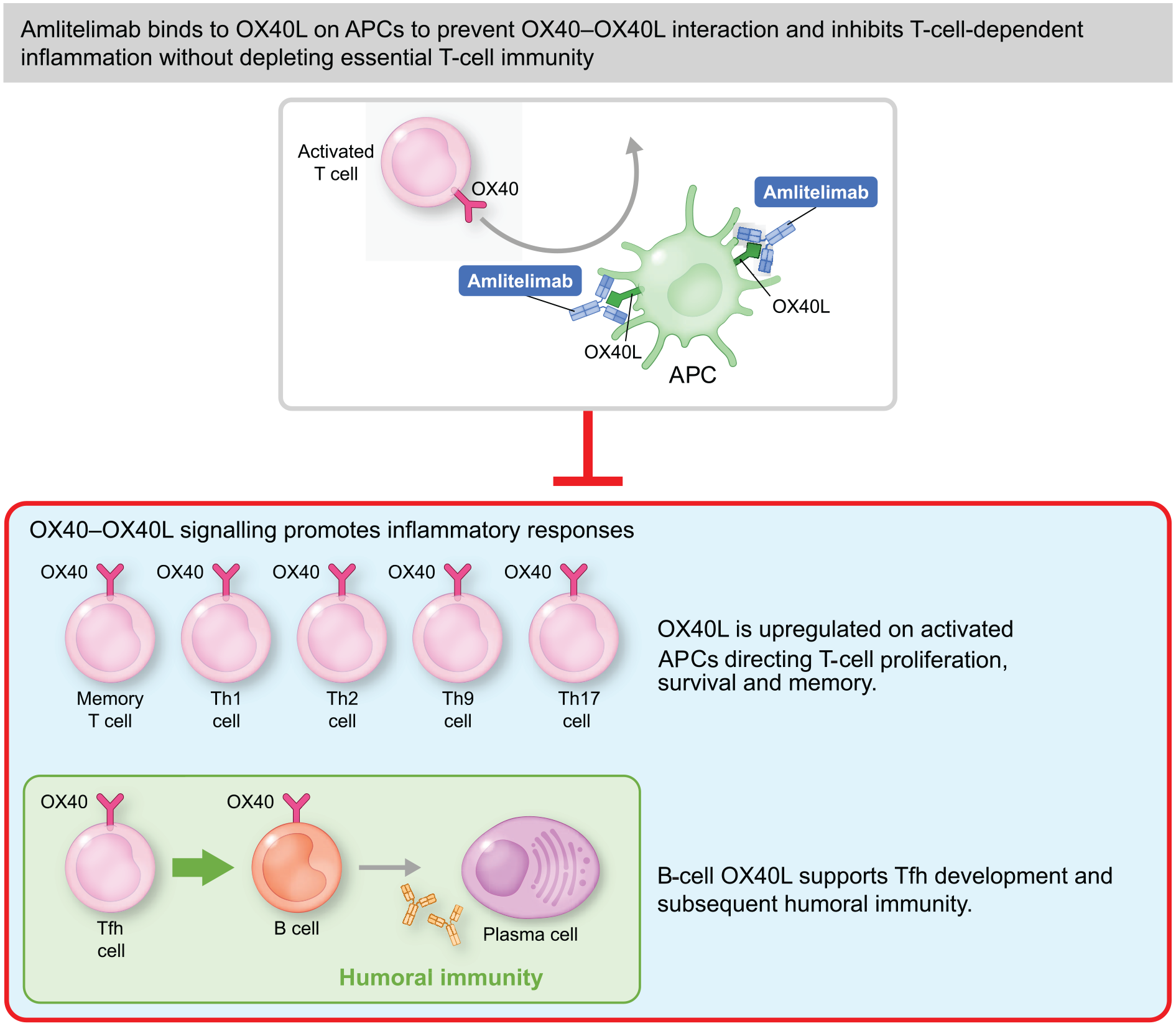
Amlitelimab detailed mechanism of action.
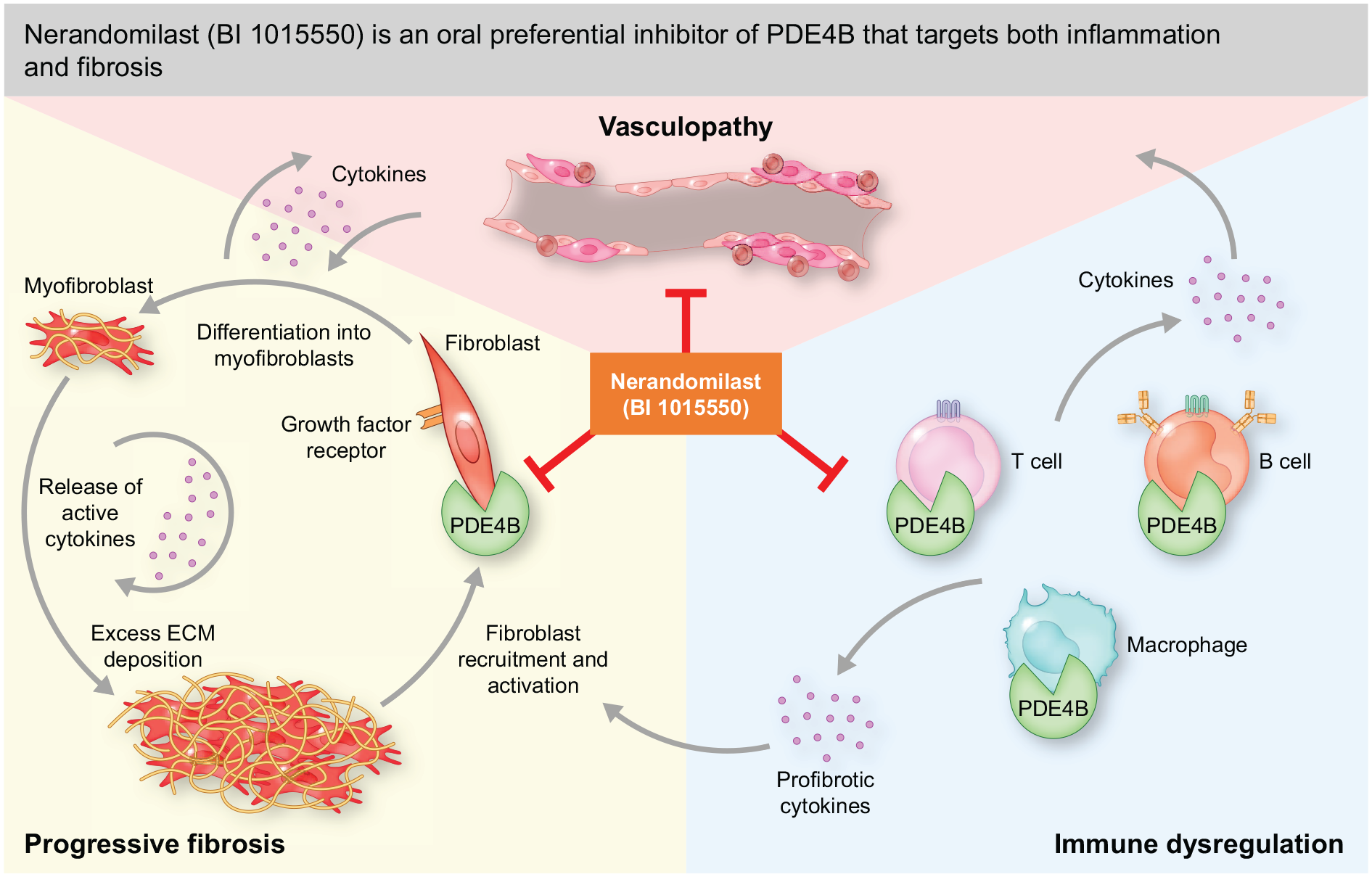
Nerandomilast detailed mechanism of action
Method
Trial design
CONQUEST is a multicentre, double-blind, randomized, placebo-controlled, Phase 2b platform clinical trial that is evaluating the efficacy, safety and pharmacodynamics of multiple IPs on pulmonary function and other endpoints in adults with early active SSc with ILD. It was designed to be an adequate, well-controlled trial consistent with the criteria described in US Food and Drug Administration (FDA) regulation 21 CFR 314.126(b). 37 Depending on regimen-specific results, data from CONQUEST could be used to design a Phase 3 trial or support the establishment of effectiveness and safety with health authorities for a particular IP.
Multiple interventional regimens are being tested in CONQUEST, each consisting of active IP versus matching placebo. Different regimens can be evaluated in parallel or asynchronously. The first IPs to be evaluated in CONQUEST will be amlitelimab and nerandomilast. See Figure 5a for an overview of the trial design.
The master protocol for CONQUEST describes the framework for the trial design population and the minimum inclusion and exclusion criteria for all regimens. The Scleroderma Research Foundation holds the investigational new drug application for the master protocol. Each IP also has a regimen-specific subprotocol, which contains intervention-specific information and defines any additional study elements and regimen-specific eligibility criteria. Only information from the master protocol is described in this article.
According to the master protocol, the trial does not have a prespecified time point for the end of the study, and enrolment into a particular regimen may start at different time points. However, regimen-specific subprotocols have predefined target enrolment numbers and end dates. Overall, the trial duration is approximately 60 weeks (4 weeks’ screening, 52-week treatment period, and a minimum of 4 weeks’ follow-up).
Patients must meet all inclusion and exclusion criteria of the master protocol and the inclusion and exclusion criteria of at least one of the two subprotocols. When two regimens are concurrent and patients fulfil the inclusion and exclusion criteria for the master protocol and both subprotocols, then the patients can be randomized to either of the subprotocols and each regimen requires only half the number of patients to be on placebo (2:1 randomization favouring active medication) (see Figure 5b). However, if some enrolled patients only fulfil the inclusion and exclusion criteria of the master protocol and one of the two subprotocols, then they are randomized to study medication or placebo (1:1 randomization) and additional numbers are needed in the active IP and placebo groups.
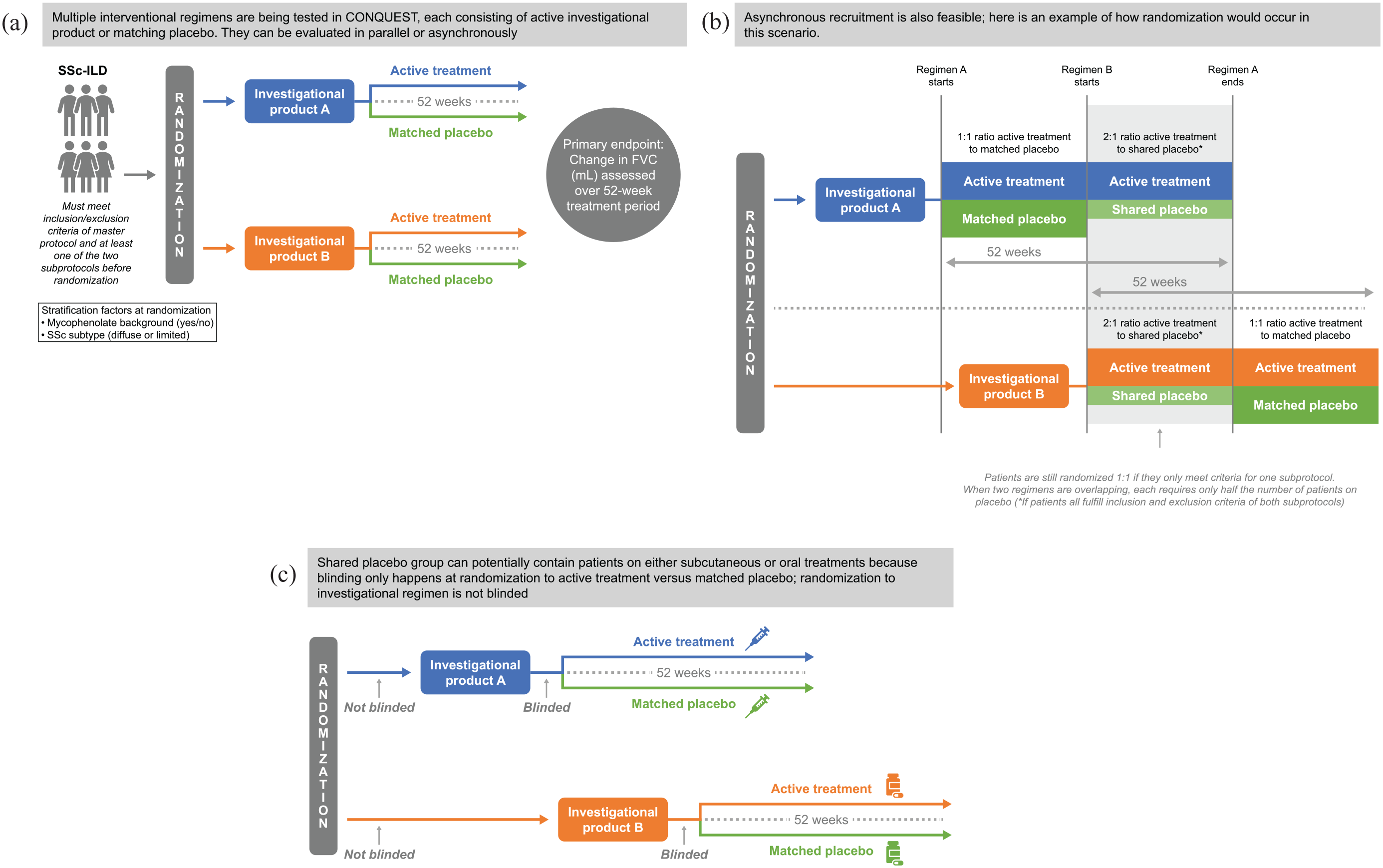
CONQUEST trial design: (a) Trial overview. (b) How sharing the placebo group reduces the number of patients needed for each regimen. (c) Randomization of oral versus subcutaneous treatments.
Trial participants, investigators and site staff are not blinded to the regimen assignment but are blinded to the assignment of patients to active product versus matching placebo. This means that the IPs can have a different frequency or mode of administration (e.g. subcutaneous versus oral; see Figure 5c).
In addition to their trial medication, trial participants can either continue receiving standard-of-care progressive scleroderma therapy (e.g. ⩽1 therapy from mycophenolate, methotrexate, or azathioprine) if the regimen is stable for ⩾6 months without dose adjustments in the last 3 months prior to randomization, or may participate without background standard-of-care therapies. If participants suffer an unexpected worsening in their disease requiring additional treatment, patients may receive rescue therapy after study start at the discretion of the investigator, but discontinuation of IP is required if any of the restricted medications are initiated during the treatment period.
Trial population
Key inclusion criteria for the CONQUEST trial include the following: male or female patients, aged ⩾18 years at the time of signed informed consent; SSc classification as defined by the 2013 American College of Rheumatology/European League Against Rheumatism criteria, 1 with SSc for ⩽5 years prior to the screening visit (including a modified Rodnan skin score (mRSS) of 10–35 for trial participants with diffuse cutaneous SSc); presence of ILD with evidence of any fibrosis on high-resolution computed tomography (HRCT) (within ⩽3 months of randomization) and at least one of the following: >10% total lung involvement on HRCT (assessed by quantitative extent of ILD in the whole lung (QILD-WL)) on central read; or elevation of acute-phase reactants at the screening visit; or progression of ILD in the previous 24 months; or forced vital capacity (FVC) ⩾45%–<80% predicted in trial participants with SSc onset (defined by first non-Raynaud’s symptom) ⩽3 years prior to the screening visit. For the full list of inclusion criteria, see Supplementary Table 1.
Key exclusion criteria include patients with clinically significant pulmonary abnormalities inconsistent with ILD on HRCT, for example, scarring due to previous active tuberculosis, sarcoidosis, lung mass or other findings unrelated to early active SSc with ILD. For the full list of exclusion criteria, see Supplementary Table 2.
An enrolment cap will apply to the limited/sine cutaneous SSc subtype, allowing for ⩽30% of limited/sine cutaneous SSc subtype trial participants for each regimen-specific subprotocol. An additional cap of ⩽20% of trial participants fulfilling only the criteria of >10% total lung involvement on HRCT (assessed by QILD-WL) will also be applied for each regimen-specific subprotocol, which may have additional inclusion/exclusion criteria.
To preserve the integrity of randomization, participants will be consented to all regimens available at that time and for which they would be eligible. Consenting participants will then be randomized to one of the regimens for which they are eligible and to either the active treatment or matching placebo group. The randomization ratio will ensure that participants have the same chance of receiving a placebo in aggregate as they have of receiving each individual active treatment. Efforts have been taken to harmonize the inclusion/exclusion criteria between regimens in order to simplify randomization and to increase trial efficiency. Randomization is also stratified by mycophenolate use and SSc subtype (limited/diffuse) to further reduce the risk of confounding. Outcomes in recipients of each experimental regimen will only be compared with outcomes in all placebo recipients who were eligible to be randomized to that experimental regimen, to preserve the integrity of randomized comparisons.
Trial objectives
The primary, secondary and exploratory objectives of the CONQUEST trial from the master protocol are described below, and trial endpoints are summarized in Table 1. Subprotocols may have additional secondary and exploratory objectives.
CONQUEST trial endpoints.

AE: adverse event; CGIC: Clinical Global Impression of Change; CGIS: Clinical Global Impression of Severity; CRISS: Composite Response Index in Systemic Sclerosis; ECG: electrocardiogram; FACIT: Functional Assessment of Chronic Illness Therapy; FVC: forced vital capacity; HCRU: healthcare resource utilization; HRCT: high-resolution computed tomography; mRSS: modified Rodnan skin score; PGIC: Patient Global Impression of Change; PGIS: Patient Global Impression of Severity; QILD-WL: quantitative interstitial lung disease – whole lung; QLF-WL: quantitative lung fibrosis – whole lung; SSc: systemic sclerosis.
The primary objective of CONQUEST is to evaluate the efficacy of the IPs compared with placebo on the change from baseline to Week 52 in FVC (mL).
Secondary objectives include the following: to evaluate the efficacy of the IPs compared with placebo, as shown by changes from baseline to Week 52 in lung involvement (measured by HRCT QILD-WL) and dyspnoea (both severity and functional limitations, measured by Functional Assessment of Chronic Illness Therapy – Dyspnoea score); to evaluate the overall treatment response (measured by the revised Composite Response Index in diffuse SSc (rCRISS) score in trial participants with diffuse cutaneous SSc at Week 52); and to evaluate the safety and tolerability of the IPs.
Exploratory objectives include assessment of the effects of IPs on other efficacy endpoints, biomarkers and healthcare resource utilization.
Statistical analysis
Determination of sample size
Power calculations for the analysis of FVC are based on the predicted effect of ~80 mL/year on FVC, an estimated value representing a clinically meaningful effect, 38 and an approximate 220 mL standard deviation, based on literature and past studies.
The trial power was estimated to be >70%, with a master-protocol-recommended sample size of a minimum of 100 patients per treatment arm. Individual subprotocols may have different sample sizes. Success will be declared at a one-sided alpha level of .025.
Stratification based on mycophenolate use and SSc subtype at screening will be used in the randomization procedure to ensure balance across regimen-specific subprotocols/treatment arms.
The targeted number of sites worldwide will be >150 centres in >25 countries. Recruitment for the trial started on 15 April 2024.
Analysis populations
The primary efficacy analysis population is the ITT population, which includes all trial participants randomized to receive an IP as well as only those placebo-control trial participants who could have been assigned to that specific IP at the time of randomization (concurrent controls). Primary analysis will not include placebo-control trial participants randomized to the control arm across other IPs before or after the randomization period of the IPs (non-concurrent non-shared controls), or patients randomized within the concurrent time in the other control arms who do not fulfil the inclusion/exclusion criteria of the specific IP (concurrent non-shared controls).
Also used for efficacy analyses, the modified ITT population is defined as all trial participants in the ITT population who adhere to IP for a minimum of 30 days and complete at least one post-baseline spirometry assessment.
Used for sensitivity analyses of the primary efficacy endpoint, the per-protocol population is defined as all trial participants in the ITT population who complete at least the Week 52 visit without any major protocol deviations.
Used for all safety analyses, the safety population is defined as all randomized trial participants who receive at least one dose of IP.
Regimen-specific subprotocols may include analysis modifications and additional analysis procedures, including interim analyses.
Ethical conduct of the trial
The trial will be conducted in compliance with the protocol and ethical principles that have their origin in the Declaration of Helsinki, the International Council for Harmonisation guidelines for Good Clinical Practice, the European Union Clinical Trials Regulation No 536/2014, and applicable regional regulations. Compliance with Good Clinical Practice provides public assurance that the rights, safety and well-being of trial participants are protected, consistent with the principles in the Declaration of Helsinki, and that the clinical trial data are credible.
The sponsor or their designee will ensure local Institutional Review Board/Independent Ethics Committee review of all appropriate trial documentation. An independent Data Monitoring Committee will meet at regular predefined intervals to review safety data, and the quality of study conduct.
Discussion
The Scleroderma Research Foundation CONQUEST platform clinical trial is the first of its kind in autoimmune diseases, using a model that was first created over a decade ago to accelerate oncology drug development.39,40
CONQUEST is designed as a Phase 2b, adequate, well-controlled trial, consistent with the criteria described in FDA regulation 21 CFR 314.126(b). 37 Depending on regimen-specific results, data from CONQUEST could be used to plan and design a Phase 3 study or may be used with health authorities to demonstrate the benefits of a particular IP outweigh its risks, either as the sole source or alongside data from other trials, or to move to Phase 3.
CONQUEST has a rigorous design intended to preserve the integrity of randomized comparisons across the CONQUEST platform. For a given regimen, the primary ITT analysis data set will include patients randomized to receive that regimen as well as any placebo-control patients who could have been assigned to that regimen when they were randomized. Hence, the primary analysis of a given drug in CONQUEST utilizes only those control-arm patients who were eligible for and underwent randomization that could have assigned them to that drug. CONQUEST does not use non-concurrent control data, even if restricting to use of contemporaneous non-current controls, as this may negatively impact the ability to make definitive conclusions regarding efficacy due to systematic differences between a drug and non-concurrent controls that could be caused by a number of factors (e.g. temporal shifts in patient characteristics, quality of trial conduct, changes in standard of care). The master protocol inclusion/exclusion criteria are adhered to as much as possible to avoid differences in subprotocol patient populations and to maximize the use of shared controls.
The use of a shared placebo-control group in the master protocol provides efficiencies over traditional trial programmes due to the more desirable randomization rates (FDA-2018-D-3292, 2022). This facilitates fewer control participants per subprotocol while maintaining statistically optimal comparison rates by sharing appropriate placebo-control patient data. Screening procedures and inclusion/exclusion criteria are provided in the master protocol to facilitate a comparable group of shared placebo-control participants at baseline across all regimens. Phase 2b trials are designed to determine if an IP has efficacy and safety data to warrant a Phase 3 trial. It is possible to obtain approval based on the results of a single adequate well-controlled Phase 2b trial, assuming the trial is conducted with few imperfections and the findings are clinically and statistically very persuasive. In general, substantiation of a drug’s effectiveness obtained with two trials is more convincing than evidence from one. Reliance on a single multicentre trial to establish substantial evidence of effectiveness is limited to special situations in which a trial demonstrates clinically meaningful and statistically persuasive effects. Depending on regimen-specific results, data could be used to support the establishment of effectiveness with health authorities.
The goal of CONQUEST is to evaluate treatments for progressive SSc. Initially, CONQUEST will focus on early active SSc with ILD, which manifests in patients as lung fibrosis, using lung function as a marker of disease progression. CONQUEST will also evaluate secondary/exploratory endpoints (e.g. rCRISS, mRSS) to test whether treatments can successfully target other organ-specific disease manifestations. In the future, the CONQUEST platform may be expanded to address other aspects of progressive SSc.
The trial design is patient centric, as it enables more patients to get access to novel medicines. However, the trial retains high statistical power for analysis of efficacy endpoints. CONQUEST will initially enrol approximately 400 patients but, by nature of the platform design, considerably fewer patients will be randomized to the placebo arm to receive standard of care. This is particularly important in rare diseases, for which patient numbers are limited and there is a need to limit the number exposed to the placebo arm, that is, patients who enter the study are more likely to be randomized to active treatment than in a traditionally designed study. This efficiency is not available in traditional trials and is one of the strengths of CONQUEST.
The CONQUEST trial design has some limitations. In addition to its complexity, the shared placebo-control group means that there is a lack of blinding to the regimen, since trial participants, investigators, and site staff are not blinded to the regimen assignment.
Patients will be enrolled across >150 centres in >25 countries. With its global network of centres dedicated to treating scleroderma, the Scleroderma Research Foundation and its pharmaceutical partners hope to improve the outlook for the scleroderma community through the framework they have designed to facilitate new drug development. The adaptable nature of the trial will allow centres to quickly pivot to testing new treatments as some treatment arms complete enrolment and new arms are added to the study. This should greatly increase the speed and decrease the cost of testing new treatments in this disease.
Conclusion
CONQUEST is a multicentre, double-blind, randomized, placebo-controlled, Phase 2b platform clinical trial evaluating the safety, efficacy, and pharmacodynamics of multiple IPs for the treatment of early active SSc with ILD. The first platform trial in the rheumatology field, CONQUEST will accelerate the development of new therapies by testing multiple IPs across a single Phase 2b platform. Amlitelimab and nerandomilast are the first molecules to be studied in CONQUEST.
Supplemental Material
sj-docx-1-jso-10.1177_23971983241278079 – Supplemental material for Design of CONQUEST, a novel, randomized, placebo-controlled, Phase 2b platform clinical trial to investigate new treatments for patients with early active systemic sclerosis with interstitial lung disease
Supplemental material, sj-docx-1-jso-10.1177_23971983241278079 for Design of CONQUEST, a novel, randomized, placebo-controlled, Phase 2b platform clinical trial to investigate new treatments for patients with early active systemic sclerosis with interstitial lung disease by Dinesh Khanna, Luke B Evnin, Shervin Assassi, Wade W Benton, Gregory Gordon, Karina Maslova, Juergen Steffgen and Toby M Maher in Journal of Scleroderma and Related Disorders
Footnotes
Acknowledgements
The authors thank all CONQUEST Steering Committee members for their valuable input. Members of the Steering Committee are: Dinesh Khanna, MD, MSc (Co-Chair), University of Michigan School of Medicine, USA; Toby Maher, MD, PhD (Co-Chair), Keck School of Medicine, USC, USA; Yannick Allanore, MD, PhD, Hôpital Cochin, France; Shervin Assassi, MD, MS, McGovern Medical School, UT Health Houston, USA; Murray Baron, MD, Jewish General Hospital, McGill University, Canada; Elana Bernstein, MD, Columbia University, USA; Vincent Cottin, MD, PhD, Louis Pradel University, Hospital Claude Bernard University, France; Francesco del Galdo, MD, PhD, University of Leeds, UK; Christopher Denton, MD, PhD, Royal Free Hospital, University College London, UK; Jeska de Vries-Bouwstra, MD, PhD, Leiden University Medical Centre, The Netherlands; Oliver Distler, MD, University Hospital Zurich, Switzerland; Robyn Domsic, MD, MPH, University of Pittsburgh, USA; Tracy Frech, MD, MS, Vanderbilt Health, USA; Jessica Gordon, MD, Hospital for Special Surgery, USA; Kristin Highland, MD, Cleveland Clinic, USA; Anna-Maria Hoffmann-Vold, MD, PhD, Oslo University Hospital, Norway; Laura Hummers, MD, Johns Hopkins Medicine, USA; Yasuhiro Kondoh, MD, PhD, Tosei General Hospital, Japan; Masataka Kuwana, MD, PhD, Nippon Medical School, Japan; Marco Matucci-Cerinic, MD, PhD, Careggi University Hospital, University of Florence, Italy; Philip Molyneaux, MD, PhD, Imperial College, UK; Virginia Steen, MD, Georgetown University, USA; Luke Evnin, PhD (Nonvoting member), Scleroderma Research Foundation; Greg Gordon, MD (Nonvoting member), CONQUEST Study Team. They also thank Hao Li from Boehringer Ingelheim, who contributed to discussions regarding the statistical aspects of the trial. The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). This was a collaborative research study where Boehringer Ingelheim was involved in the study design but was not the regulatory sponsor. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as it relates to Boehringer Ingelheim substances, as well as intellectual property considerations. Cindy Macpherson, PhD of Nucleus Global provided editorial support, which was contracted and funded by the Scleroderma Research Foundation.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: D.K., via his institution, has received industry-academic funding from Boehringer Ingelheim, BMS, Merck and GlaxoSmithKline R&D; and consultancy fees and/or on Scientific Advisory Board from AbbVie, Amgen, Argenx, AstraZeneca, Boehringer Ingelheim, BMS, Galapagos, GlaxoSmithKline, Merck, Mirador, Mitsubishi Tanabe Pharma Corporation, Novartis, Roche, Sanofi-Aventis and Zura Bio. L.B.E. currently represents MPM Capital on the Board of Directors for each of Photys Therapeutics, Trishula Therapeutics, Frontier Medicines, Redona Therapeutics and Umoja Biopharma. He is Chairman of the Board of Werewolf Therapeutics and owns stock in Eicos Sciences, an affiliate of CiVi Biopharma. S.A. declares grant support to his institution from Scleroderma Research Foundation, Boehringer Ingelheim, aTyr and Janssen, as well as personal consultancy fees from AbbVie, aTyr, AstraZeneca, Boehringer Ingelheim, CSL Behring and Merck. W.W.B. is a paid consultant of the Scleroderma Research Foundation. G.G. is an employee of the Scleroderma Research Foundation. K.M. is an employee of Sanofi. J.S. is an employee of Boehringer Ingelheim. T.M.M., via his institution, has received industry-academic funding from AstraZeneca and GlaxoSmithKline R&D; and consultancy or speaker fees from AstraZeneca, Bayer, Boehringer Ingelheim, BMS, CSL Behring, Endeavour, Fibrogen, Galapagos, Galecto, GlaxoSmithKline, IQVIA, Merck, Pliant, Pfizer, Qureight, Roche, Sanofi-Aventis, Structure Therapeutics, Trevi and Vicore. He is supported by a British Lung Foundation Chair in Respiratory Research (C17-3).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: Funding for the clinical trial was provided by Sanofi and Boehringer Ingelheim. Scleroderma Research Foundation received support from Sanofi and Boehringer Ingelheim to develop the protocol with Medpace and conduct initial feasibility work. Nucleus Global provided editorial support for the development of the manuscript, which was contracted and funded by the Scleroderma Research Foundation.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.