
Abstract
We present a case series of four patients with systemic sclerosis and skeletal myopathy. While idiopathic inflammatory myopathies, or myositis, are thought to be the most common type of muscle disease seen in systemic sclerosis, we highlight four cases where unique clinical findings and careful assessment ruled out myositis mimics. Key diagnostic tools that can be helpful for clinicians to diagnose a neuromuscular disease are also detailed in this report.
Introduction
Skeletal myopathy is an important, yet underappreciated manifestation of systemic sclerosis or scleroderma (SSc) that leads to poor outcomes such as disability and mortality.1,2 When myopathy is suspected, careful evaluation with physical exam and key diagnostic tools can be helpful to determine the etiology. The purpose of this case series is to highlight neuromuscular disorders that can mimic the myositis and/or fibrotic muscle disease typical of SSc.
This is a retrospective case series of four patients with SSc and proximal muscle weakness who were initially believed to have an idiopathic inflammatory myopathy (IIM), but key diagnostic tools proved to be instrumental in diagnosing other neuromuscular diseases. All four patients met the 2013 ACR/EULAR classification criteria for SSc 3 and consented to be a part of an Institutional Review Board–approved Johns Hopkins Scleroderma Registry.
Description of case series
Patient 1 was a 36-year-old female with history of diffuse SSc and U1-RNP positivity who presented with proximal muscle weakness with a maximum creatine kinase (CK) of 500 U/L. Muscle magnetic resonance imaging (MRI) demonstrated edema of the pelvic muscles. Electromyography (EMG) showed non-irritable myopathy. Muscle biopsy did not demonstrate any inflammation or necrosis. Commercial myositis autoantibody testing was negative. With the lack of biopsy findings to support an inflammatory myopathy, further studies for myasthenia gravis (MG) were conducted due to her progressive muscle fatigue. Single fiber EMG of the left frontalis muscle showed abnormal jitter and blocking, a classic finding for a neuromuscular transmission defect. Acetylcholine receptor binding (AChR) antibody was also positive at 0.91 (normal < 0.30 nmol/L) consistent with MG. She was treated with pyridostigmine, mycophenolate mofetil (MMF), and intravenous immunoglobulin (IVIG) with marked improvement in her symptoms.
Patient 2 was a 58-year-old woman with a history of diffuse SSc and prior diagnosis of non-ischemic cardiomyopathy at the age of 41. She was treated with MMF 3 g/daily for her skin disease and developed 1 year of gradual proximal muscle weakness prominent in the neck flexor and arm abductors. Her maximum CK was 990 U/L. EMG demonstrated a non-irritable myopathy. Commercial myositis autoantibody testing was negative. Repetitive nerve stimulation test was normal. Muscle MRI of the left humerus did not show muscle edema. Muscle biopsy of the left deltoid showed mild non-specific myofiber atrophy without inflammation or fibrosis. Owing to a strong family history of familial cardiomyopathy, she was referred for genetic testing which showed a pathogenic mutation in MYH7 (myosin heavy chain 7). The MYH7 gene is important in producing the protein beta-myosin heavy chain, which is found in cardiac and type I skeletal muscle fibers. Type I fibers are important in resisting fatigue and keeping posture (neck muscle strength). 4 She was treated for her cardiomyopathy, and physical therapy was initiated for her weakness.
Patient 3 is an 82-year-old woman with limited SSc and anti-centromere positivity who developed weakness in arms and around her scapulae over several years. Physical exam revealed neck flexor, arm abductor, and hip flexor weakness. She also had scapular winging and mild facial weakness on exam. Maximum CK was 128 U/L. A bilateral thigh MRI showed mild muscle edema. EMG/NCS showed non-irritable myopathy. Commercial myositis autoantibody testing was negative. Muscle biopsy was not done due to the prominent physical exam findings concerning for facioscapulohumeral dystrophy (FSHD). Genetic testing confirmed FSHD deletion mutation on chromosome 4q35, which is inherited as an autosomal dominant disorder.
Patient 4 is a 78-year-old female with history of limited SSc and centromere positivity who presented with mild proximal muscle weakness over 10 years that began to get worse with exercise. She also reported dyspnea on exertion and developed a cardiomyopathy. There was no evidence of pulmonary hypertension or interstitial lung disease. Maximum CK was 176 U/L. EMG showed a non-irritable myopathy. Muscle MRI was not completed. Commercial myositis autoantibody testing was negative. Muscle biopsy showed features of a mitochondrial myopathy with too numerous to count COX-negative fibers without any ragged red fibers. She was referred to the neuromuscular department for genetic testing.
Conclusion
Our case series of four patients with SSc discovered to have non-scleroderma-related etiologies of muscle weakness included MG, familial cardiomyopathy with skeletal muscle involvement, FSHD, and mitochondrial myopathy mimicking scleroderma-related muscle disease. The prevalence of skeletal myopathy in SSc have been reported to be 15% in a systematic review, 5 with overlap IIMs being the most common etiology for proximal muscle weakness. It is unknown how often distal weakness occurs in patients with skeletal myopathy, but most studies defining the clinical presentation of myopathy in SSc have reported the pattern of weakness is commonly proximal in distribution.6 –8 In a recent retrospective study of skeletal myopathy at our center from 1990 to 2021, the frequency of myopathy as defined as proximal weakness and/or elevated CK with at least one abnormal diagnostic testing revealed that there was 17% (678 of 3923). 9 These four cases reported here were noted in the past 15 years due to its unexpected diagnosis when undergoing myopathy evaluation at our institution. Our study highlights why it is important to entertain a broad differential diagnosis especially (Figure 1) when there is a family history of neuromuscular diseases, lack of response to immunosuppression, or compelling exam findings. For example, Patient 1 had progressive fatigable weakness that led to a diagnosis of MG, and Patient 3 had scapular winging, a hallmark feature of FSHD. Key diagnostic tools such as electrodiagnostic testing and muscle biopsy can be useful in the work-up of SSc-associated myopathy (Table 1). These cases highlight the importance of identifying unique physical exam features or clinical symptoms of muscle disease that does not fit the classic clinical presentation for myositis.
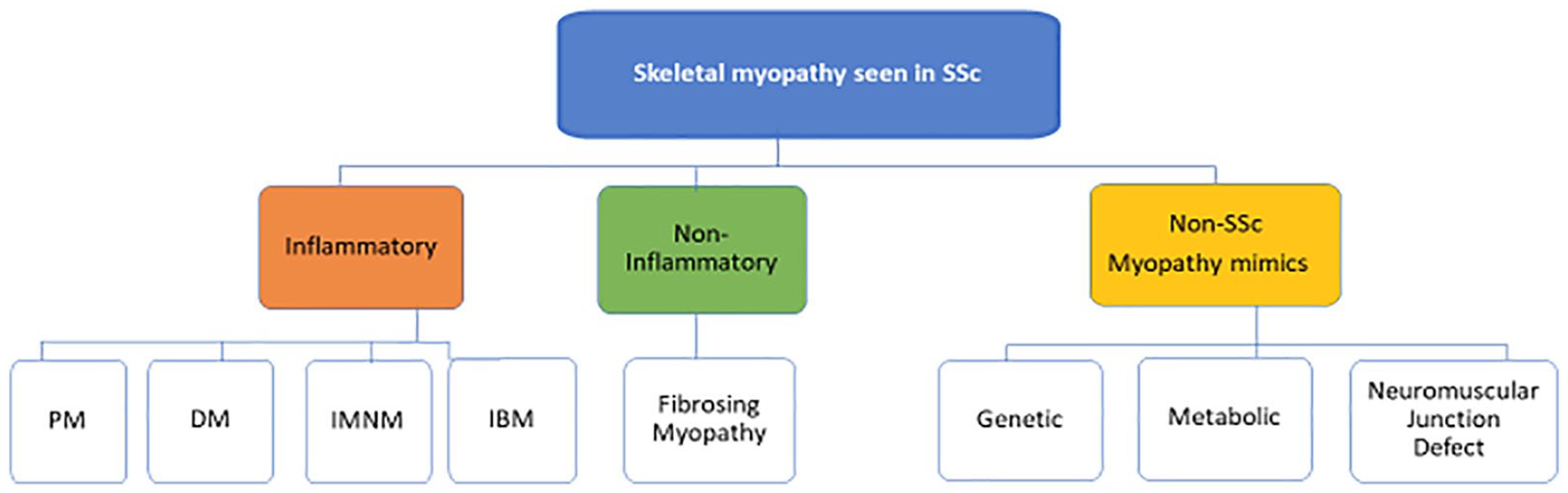
The spectrum of skeletal myopathy in systemic sclerosis.
Clinical characteristics of patients with scleroderma and neuromuscular diseases highlighting key clinical tools for diagnosis.
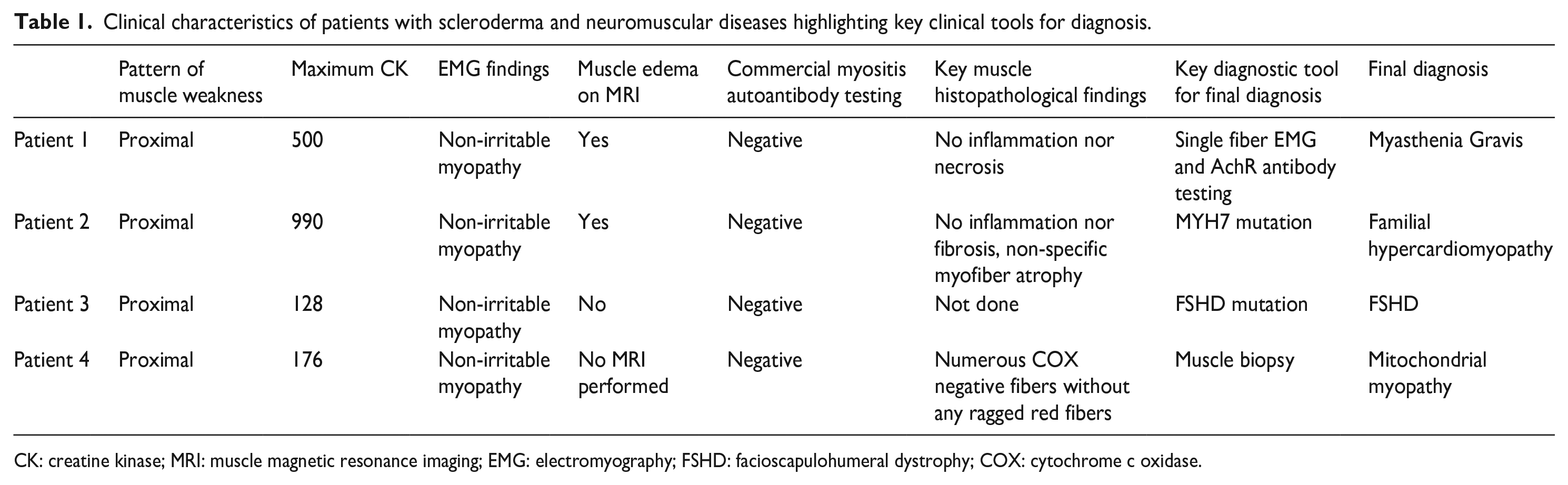
CK: creatine kinase; MRI: muscle magnetic resonance imaging; EMG: electromyography; FSHD: facioscapulohumeral dystrophy; COX: cytochrome c oxidase.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support was provided by NIAMS (K23AR073927).