
Abstract
Objective:
To describe the efficacy and safety in all patients with systemic sclerosis–associated pulmonary arterial hypertension who started selexipag between 09-2016 and 06-2018 in two pulmonary arterial hypertension expert centers.
Methods:
All patients with systemic sclerosis–associated pulmonary arterial hypertension diagnosed by right heart catheterization and treated with selexipag were included. Every 12 weeks, treatment effect was assessed by (1) the opinion of the expert team and (2) the abbreviated risk assessment, consisting of functional class, six-minute walking distance, and N-terminal prohormone of brain natriuretic peptide level at baseline and during follow-up. Side effects and adverse events were registered.
Results:
We included 13 systemic sclerosis–associated pulmonary arterial hypertension patients, 10 patients were female, median age (interquartile range) of 68 (58–75) years, median systemic sclerosis disease duration of 7.4 (4.7–13.5) years, and median pulmonary arterial hypertension duration of 4 (2.5–7.5) years. Two patients discontinued selexipag within 4 weeks due to side effects. The remaining 11 patients had a median follow-up duration of 48 (interquartile range = 24–72) weeks. Two patients died (one pulmonary arterial hypertension–related, the other systemic sclerosis–related). According to the expert team, 8 of 11, 9 of 10, and 5 of 7 patients stabilized or improved at 12, 24, and 48 weeks, respectively. According to the abbreviated risk assessment at study end, 3 of 11 patients had 1 low-risk criterion. No previously unrecorded side effects were reported.
Conclusion:
Adding selexipag to background therapy in a high-risk cohort of systemic sclerosis–associated pulmonary arterial hypertension patients provided sustained stabilization of symptoms with an acceptable safety profile. Improvement was reached in only two of our patients. Further research should focus on systemic sclerosis–associated pulmonary arterial hypertension patients treated with multiple targeted treatments, preferably these patients should be prospectively followed in international registries.
Introduction
Pulmonary arterial hypertension (PAH), diagnosed by right heart catheterization, has a prevalence of 8%–12% in systemic sclerosis (SSc) patients and may occur at any stage. 1 As diagnosing SSc with mild or no cutaneous involvement can be difficult for nonexperts, PAH occurrence can be among the first symptoms of SSc. The 3-year survival rate of PAH associated with SSc (SSc-PAH) is estimated to be only 52%, despite the available targeted treatments. 2 Current treatment strategies are based on initial evaluation of severity and prognostic risk, as well as subsequent response to treatment.3,4 Risk status for mortality of idiopathic-PAH patients can be assessed by a comprehensive tool described by the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) guidelines.3,5 The number of low-risk criteria at baseline and during the first follow-up discriminates the risk of death or lung transplantation and enables classification of patients as low (<5%), intermediate (5%–10%), or high risk (>10%) for an estimated 1-year mortality. However, as SSc-PAH is a more progressive disease, treatment is focused on delay of progression and on achieving as many low-risk criteria as possible. Until the registration of the oral prostacyclin receptor agonist (PGI2) selexipag in 2016, treatment targeting the prostacyclin pathway was only available as continuous intravenous, subcutaneous (SC), or inhalation therapy. Although these medications were registered and recommended for high-risk SSc-PAH patients, due to practical barriers of self-administering and possible complications of parenteral medication, patients often were not able to initiate or continue with these treatments. 3 With the availability of selexipag, treatment with prostacyclines has become feasible for more SSc-PAH patients. In this article, we report on the efficacy and safety of selexipag in SSc-PAH patients in a real-life case series.
Case description
We included all patients treated with selexipag at the pulmonary hypertension (PH) expert centers of the Radboud University Nijmegen Medical Centre and the Oslo University Hospital between 09-2016 and 06-2018. Inclusion criteria were (1) fulfillment criteria for SSc, (2) age >18 years, (3) PAH (WHO group 1) diagnosed by right heart catheterization. 3 Clinical and demographic data were prospectively collected, including New York Heart Association functional class (NYHA), six-minute walking distance (6MWD), N-terminal prohormone of brain natriuretic peptide (NT-pro-BNP), and co-medication during follow-up. Both SSc and PAH disease duration were calculated from the time of diagnosis until start of selexipag. Patients were categorized as having no, limited, or extensive interstitial lung disease (ILD) by combining the pulmonary function test results and high-resolution computed tomography scan results as described by Goh et al. 6 Side effects and adverse events were recorded weekly during titration and every 3 months thereafter. Data on reason for discontinuation, treatment alterations, hospitalization, and cause of death were collected from the patient electronic medical file. Follow-up was performed every 3 months according to ESC/ERS guidelines recommendations and local institution protocol, and consisted of evaluating symptoms, clinical signs of right heart failure, syncope, NYHA class, 6MWD, NT-pro-BNP, imaging, echocardiography, and exercise testing, if applicable. 3 We defined treatment effect according to the expert team as “improvement,” “stabilization,” or “deterioration” taking into account all these parameters. Treatment effect was also evaluated by the abbreviated risk assessment, 5 which consists only of noninvasive parameters. Low-risk criteria are defined as NYHA functional class I or II, 6MWD >440 m, and NT-pro-BNP <300 pg/mL recorded at treatment initiation and during follow-up.
Thirteen SSc-PAH patients were included. All patients were classified as limited cutaneous systemic sclerosis (LcSSc), 10 patients were female. The median age was 68 (interquartile range (IQR) = 58–75) years, median disease duration for SSc at initiation of selexipag was 7.4 (IQR
Patient characteristics at baseline and during follow-up.
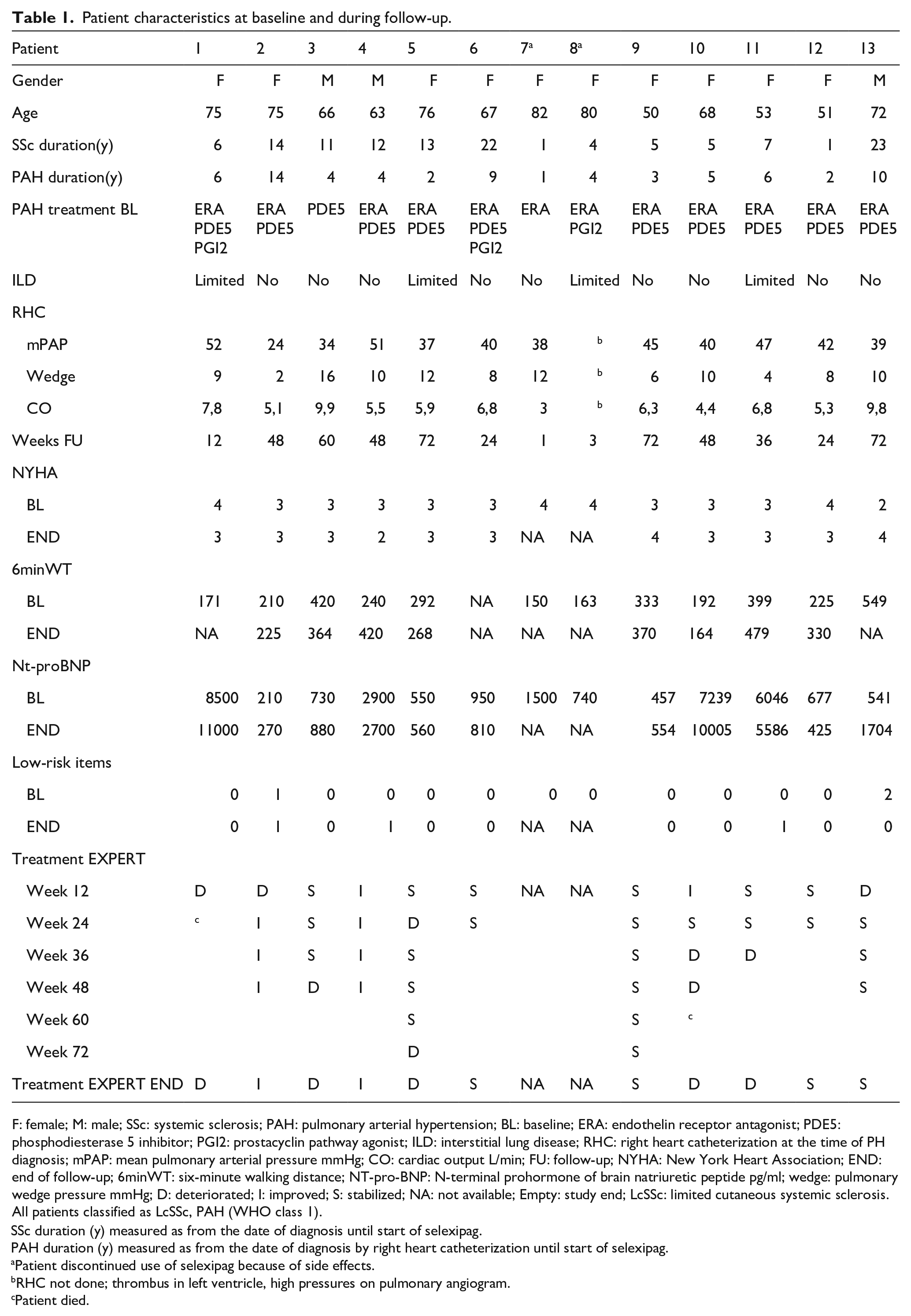
F: female; M: male; SSc: systemic sclerosis; PAH: pulmonary arterial hypertension; BL: baseline; ERA: endothelin receptor antagonist; PDE5: phosphodiesterase 5 inhibitor; PGI2: prostacyclin pathway agonist; ILD: interstitial lung disease; RHC: right heart catheterization at the time of PH diagnosis; mPAP: mean pulmonary arterial pressure mmHg; CO: cardiac output L/min; FU: follow-up; NYHA: New York Heart Association; END: end of follow-up; 6minWT: six-minute walking distance; NT-pro-BNP: N-terminal prohormone of brain natriuretic peptide pg/ml; wedge: pulmonary wedge pressure mmHg; D: deteriorated; I: improved; S: stabilized; NA: not available; Empty: study end; LcSSc: limited cutaneous systemic sclerosis.
All patients classified as LcSSc, PAH (WHO class 1).
SSc duration (y) measured as from the date of diagnosis until start of selexipag.
PAH duration (y) measured as from the date of diagnosis by right heart catheterization until start of selexipag.
Patient discontinued use of selexipag because of side effects.
RHC not done; thrombus in left ventricle, high pressures on pulmonary angiogram.
Patient died.
In total, three SSc-PAH patients did receive parenteral or inhaled PGI2 treatment before the start of selexipag. Patients 1 and 8 switched from iloprost nebulization to selexipag on their own request to improve quality of life, as nebulization multiple times a day was time-consuming. Patient 6 requested to switch from trepostinil SC to selexipag because she experienced pain during administration. In all three cases, PGI2 was faded out gradually during titration period.
Two patients discontinued selexipag within 4 weeks due to side effects, and the remaining 11 patients had a median follow-up duration of 48 (IQR = 24–72) weeks. Dosage of selexipag was titrated according to tolerability, ranging from 800 to 1600 µg twice daily. Medium maintenance dosage was reached in three patients, high maintenance dosage in eight patients. Maximum tolerable dosage was reached after a median of 13 weeks, ranging from 7 to 41 weeks. At study end, with a median follow-up duration of 48 weeks, 2 patients (18%) improved, 4 patients (36%) stabilized, and 5 patients (46%) deteriorated. Of these 5 patients that deteriorated, 3 (60%) initially stabilized for 12 to 60 weeks. At the start of selexipag treatment, one patient had one low-risk criterion, and one patient had two low-risk criteria. At study end, 3 (27%) of the 11 patients had 1 low-risk criterion; none had 3 low-risk criteria (Table 1). During follow-up, 2 SSc-PAH patients died. One patient developed right ventricular failure, 12 weeks after the initiation of selexipag. The other patient developed uncontrollable gastro-intestinal bleeding due to angiodysplasias (55 weeks after the start of selexipag). All patients experienced side effects during titration period, most common were headache, nausea, and jaw pain. During maintenance dosage, all side effects subsided or diminished in intensity. Side effects did not differ between patients with mild ILD and without ILD. Two patients discontinued selexipag due to intolerable side effects. The first patient experienced severe dyspnea and peripheral edema 1 week after initiation. The second patient experienced severe headache, nausea, jaw pain, diarrhea, and abdominal pain 3 weeks after initiation, caused by overdosing of prescribed selexipag. In both cases, selexipag was discontinued, resulting in resolving of complaints within 1 week.
In this case series, we describe a group of 13 SSc-PAH patients with adverse prognosis who started with selexipag in addition to background therapy followed at two tertiary expert centers for PH. Our study shows sustained stabilization of symptoms for the majority of patients during multiple follow-up moments ranging from 12 to 60 weeks, and selexipag was well tolerated past the titration phase. We cannot confirm improvement as reported in the previous studies. Furthermore, the patients with both PAH and limited ILD responded worse on selexipag, probably reflecting a more severe disease.
The subanalysis of the pivotal selexipag trial describing SSc-associated and connective tissue disease–associated PAH patients showed a reduction of the primary composite endpoint (morbidity/mortality) of 44% and 41%, respectively, by adding selexipag versus placebo.7–9 Compared to the PAH patients included in these subanalyses, our patients have a higher risk of mortality; that is, older age, longer duration of PAH and a higher NYHA class at the start of selexipag. 8 Moreover, 12 of our 13 patients received double background therapy. Three patients were switched from either iloprost nebulization or trepostinil SC to selexipag on their own request after consultation of the expert team. These patients were not expected to improve from this switch; treatment goal was to improve their quality of life.In this study, we used two outcome measures, expert opinion and noninvasive low-risk criteria, which show discrepancy. Based on expert opinion, patients 2 and 4 improve at study end. Based on expert and noninvasive parameters combined, only patient 4 improves. Also patient 13 stabilizes according to expert opinion, but this is not reflected in the amount of noninvasive risk criteria as there is a deterioration of NT-pro-BNP. This discrepancy is due to the fact that the expert opinion is not only based on the noninvasive parameters but takes into account all the available determinants of prognosis as described in the risk assessment of the ESC/ERS guidelines for that patient. 3
A recent large multicenter cohort study of the French PAH registry including newly diagnosed SSc-PAH patients showed that only a minority of patients (28%) achieved 3–4 low-risk criteria after initial treatment, and long-term survival was still poor. 10 In our study, only three patients reach one low-risk criterion at study end. It should be considered that reaching low-risk status may not be realistic and achievable in SSc-PAH patients with advanced disease, severe co-morbidities, and older age such as in our cohort. 3 Another acceptable outcome may also be prevention of progression in SSc-PAH.
The strength of this study is the real-life report on the use of selexipag in clinical practice in patients with SSc-PAH with adverse prognosis. Our study however has some important limitations such as the small sample size. All patients were recruited from two PH expert centers, which could have resulted in a selection bias. However, the care for patients with rare diseases such as SSc-associated PAH is preferably organized in expert centers. Recruitment and inclusion of these patients is inevitably performed at these facilities. Also, no hemodynamic follow-up data were available for our patients, as is recommended by the guidelines in case of clinical deterioration. In clinical practice for these patients, no hemodynamic monitoring was performed as there were no clinical consequences for the adaptation in treatment regime. Finally, the abbreviated risk assessment tool used to evaluate treatment effect in addition to the ESC/ERS guidelines is not validated to date in SSc-PAH patients, necessitating to interpret our findings with caution.
Conclusion
In this study, we found that treatment with selexipag in SSc-PAH patients in real life is safe and can attribute to stabilization. Improvement was reached in only two of our patients. Further research should focus on gathering more insight in real-life prospectively followed cohorts treated with multiple targeted treatments. Our findings suggest adding selexipag in SSc-PAH patients with poor prognosis, who are unwilling or unable to pursue intravenous or subcutaneous prostacyclin targeted therapy, should be considered in clinical practice.
Footnotes
Acknowledgements
The authors are grateful to all the patients at their respective institutions who made this study possible. All authors contributed to the final article preparation.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: H.F. reports a speakers fee from GSK, and received travel bursaries from GSK and Actelion outside the submitted work. F.H.J.H. reports speakers fees from Amgen, Boehringer Ingelheim, Novartis, and consulting fees from AbbVie, Actelion, Biogen, BMS, Celltrion, Corbus, Eli-Lilly, Mundipharma, Pfizer, and Sanofi-Genzyme outside the submitted work. A.P.D. reports an unrestricted educational grant for PhD student, a speakers fee, and a consultation free from Actelion outside the submitted work. A.M.H.-V. reports research support, consulting fees, and a speakers fee from Boehringer Ingelheim and consulting and speakers fees from Actelion outside the reported work. M.C.V. reports an unrestricted educational grant, research support, speakers fees, and consulting fees (advisory board) from Actelion, research support from Ferrer, speakers fee and consulting fees (advisory board) from Boehringer Ingelheim, and a speakers fee from Roche outside the submitted work. The other authors declare no conflicts of interest.
Ethical approval
The study was approved in Oslo by The Regional Committee of Health and Medical Research Ethics in South-East Norway, Research Protocol No. 2016/119. Due to the observational nature of this study, no ethical review was needed according to Dutch law and regulations. The study complies with the Declaration of Helsinki.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Notation of prior abstract publication/presentation
Annual European Congress of Rheumatology EULAR2019 Madrid, Spain, 12–15 June 2019 abstract and poster presentation.