
Abstract
Pulmonary veno-occlusive disease is a rare cause of pulmonary hypertension in patients with systemic sclerosis that can be misclassified as pulmonary arterial hypertension. Differentiation between pulmonary veno-occlusive disease and pulmonary arterial hypertension is challenging because of the similar clinical picture. Nevertheless, discrimination is important because pulmonary veno-occlusive disease has a worse prognosis. Vasodilators including phosphodiesterase type 5 inhibitors and endothelin receptor antagonists should be started with caution and often in combination with diuretics to prevent pulmonary edema.
Keywords
Introduction and background information
Systemic sclerosis (SSc) is a chronic and severe auto-immune disease characterized by small vessel arterial vasculopathy, and fibrotic and inflammatory processes. Pulmonary complications including pulmonary arterial hypertension (PAH) are a leading cause of death in patients with SSc. Pulmonary veno-occlusive disease (PVOD) is a rare cause of pulmonary hypertension (PH) that can be misclassified as PAH. It is challenging to distinguish PAH from PVOD as the clinical picture is quite similar with dyspnea and signs of right heart failure. 1 PAH and PVOD show specific biological and morphologic similarities and, therefore, both conditions are considered within one large spectrum of diseases.2,3 PAH is mostly described as constrictive remodelling of the pre-capillary pulmonary vasculature, where the hallmarks of PVOD are capillary remodelling and extensive pulmonary venous lesions. The aetiology of PVOD is still largely unknown. Mutations (eukaryotic initiation factor 2 alpha kinase 4 gene) and different risk factors (genetic, auto immune disorders, organic solvent exposure) seem to play a role in the development of PVOD. 2 Histologically, PVOD is characterized by an obliterative fibrotic vasculopathy, predominantly affecting smaller branched vessels of the venous pulmonary tree. The affected small arterioles but primarily the affected venules show intimal proliferation, collagen matrix deposition and disorganized smooth muscle hypertrophy in patients with PVOD. 4 As a result, the lumina of these vessels will narrow or occlude. In PVOD, alveolar capillaries are typically engorged and dilated due to downstream obstruction, and frank angioproliferation may even occur. Both PAH and PVOD lead to an increase in pulmonary vascular resistance (PVR) and right ventricular failure.
In SSc, microvascular damage is characterized by endothelial cell injury and apoptosis followed by progressive intima proliferation and vessel narrowing and obliteration. It has been suggested that the vasculopathy in SSc may involve post-capillary pulmonary venules more often than previously expected resulting in a PVOD-like vasculopathy. 5 Patients with SSc indeed seem to have a high prevalence of PAH/PVOD (8%). 6
In the next section, the case of a 45-year-old female recently diagnosed with SSc and PVOD will be presented.
Case description
We present the case of a 45-year-old female, who was referred to the Combined Care in Systemic Sclerosis (CCISS) programme of the Leiden University Medical Center (LUMC) 7 for second opinion because of severe dyspnea. She had been diagnosed with limited cutaneous SSc in 2017 (based on Raynaud’s phenomenon, skin thickening involving the fingers and the face and starting in 2017, abnormal nailfold videocapillaroscopy with dilatations and haemorraghes and presence of anticentromere antibodies). The patient presented in our clinic with rapidly progressive dyspnea and worsening of exercise tolerance over the last 8 months despite treatment with mycophenolate mofetil (MMF) since 4 months. MMF was started by her treating rheumatologist prior to the first visit to our clinic for skin involvement with progressive dyspnea. Other co-medications included hydrochlorothiazide, enalapril (by reason of hypertension) and nifedipine (for Raynaud’s). On clinical examination, we observed shortness of breath on minimal exertion, pitting scars, modified Rodnan Skin Score (mRSS) of 3/51, calcinosis and telangiectasia on her fingers. Pulmonary auscultation was normal, and peripheral oedema was present around the ankles. Oxygen saturation in ambient air (peripheral capillary oxygen saturation (SpO2)) and in resting position was 97%. She had a sinus tachycardia at rest (116/min) with a normal blood pressure (110/79 mmHg). Electrocardiography showed sinus tachycardia, but no arrhythmias, and no conduction or repolarisation abnormalities. Six-minute walk test (6-MWT) resulted in a maximal walking distance of 123 m with a SpO2 of 97% at start (heart rate 115/min); after 1 min of walking SpO2 dropped to 82%; SpO2 restored to 100% with oxygen suppletion and in rest; after restart SpO2 again rapidly dropped to 84% and the test was ended. Complete laboratory examinations including erythrocyte sedimentation rate (ESR) (29 mm/h), N-terminal pro-B-type natriuretic peptide (NT-proBNP) (32.0 ng/L), creatine kinase (CK) (58 U/L) and troponine T (4 ng/L) levels were normal. Pulmonary function testing revealed normal spirometry with decreased diffusion capacity for carbon monoxide (DLCO; 38% of predicted). Transthoracic echocardiography showed no signs of diastolic dysfunction; maximum tricuspid incompetence (TI) gradient was 19 mmHg; and left ventricular function (ejection fraction 45%) was decreased with diffuse hypokinesia. Most importantly, there were no direct or indirect signs of PH. Previous high resolution chest tomography (HRCT) did not reveal any interstitial lung disease (ILD). Given unexplained and severe dyspnea on exertion, HRCT was repeated in our clinic. This showed centrilobular ground glass opacifications (partially confluent), some septal lines, and enlargement of mediastinal and hilar lymph nodes (Figure 1). Given the unexplained and severe dyspnea, without clear signs of P(A)H on echocardiography, first a computed tomography angiography (CTA) of the thorax was made which excluded pulmonary embolisms but showed dilatation of the pulmonary artery (Figure 2), lymphadenopathy, mild dilatation of the right ventricle and of the right atria and in addition some flattening of the septum. A ventilation/perfusion scan did not show any perfusion or ventilation defects. Cardiopulmonary exercise testing showed a VO2max of 56% of predicted and AaDO2 increased from 5.5 kPa at rest to 9.9 kPa during exercise. To exclude shunting at a cardiac level as a cause of hypoxaemia on minimal exertion, a contrast echocardiography was performed which was normal. Cardiac magnetic resonance imaging was unobtainable due to morbid obesity (body mass index (BMI) = 43) of the patient. Despite no previous transthoracic echocardiographic findings in favour of PH, but based on the strong clinical suspicion of PAH/PVOD (without any other explanation for the dyspnea), a right heart catheterization was performed. This revealed a pulmonary artery pressure (PAP) of 56/27 mmHg with a mean pulmonary artery pressure (mPAP) of 39 mmHg, pulmonary capillary wedge pressure (PCWP) was 13 mmHg, cardiac output (CO) 8 L/min, cardiac index value 3.3 L/min/m2, right atrial pressure (RAP) 10 mmHg and PVR was 3.3 Wood Units.
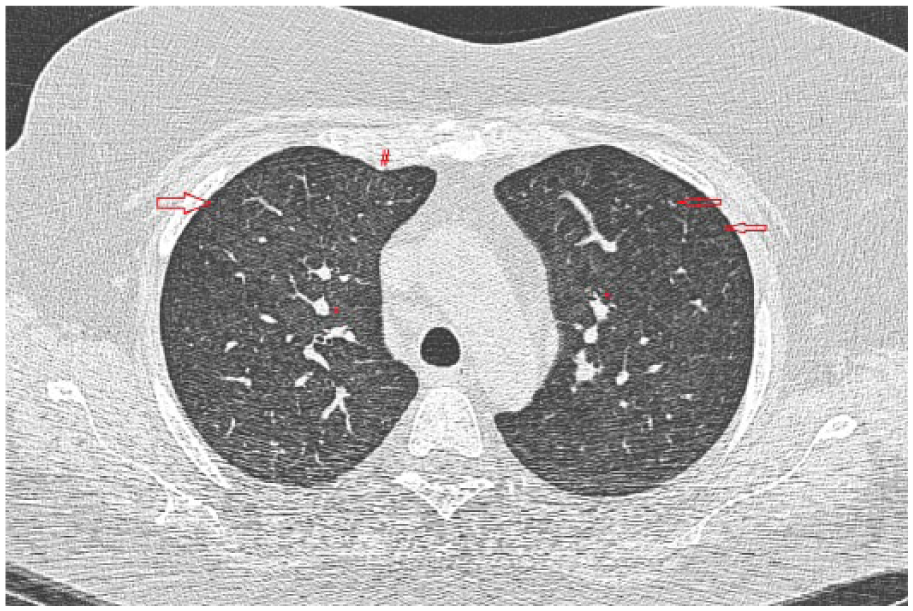
HRCT with centrilobular located ground glass opacities in the peripheral lung parenchyma (arrows). The ground-glass opacities are more confluent to the central part of the lung. The pulmonary arteries (*) are enlarged and some subtle septal lines (#) are seen in the right upper lobe.
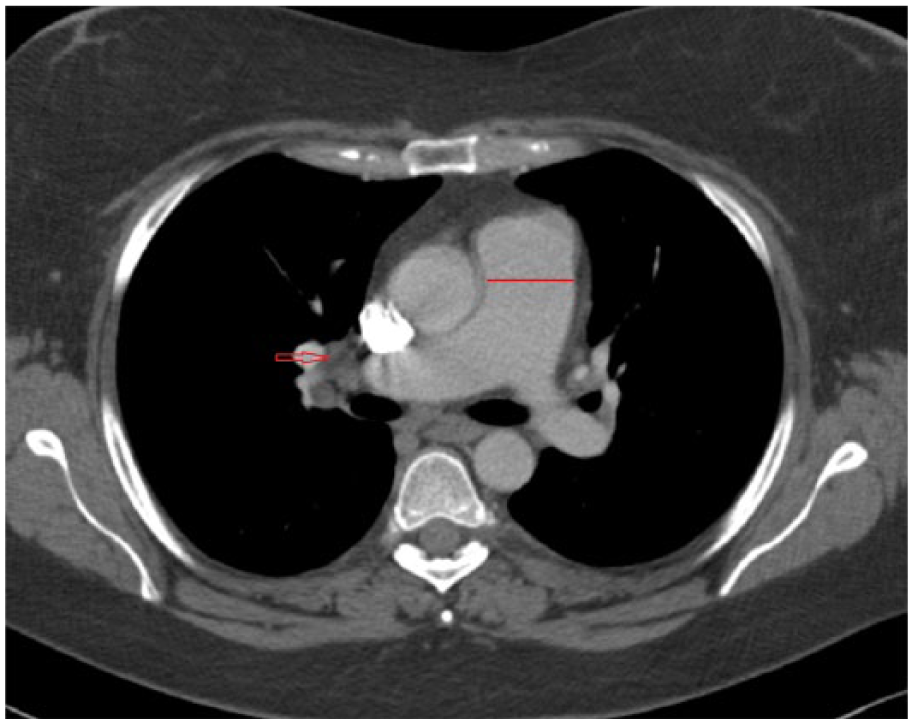
CTA of the thorax showing dilatation of the pulmonary trunk and mediastinal lymphadenopathy on the right side.
Diagnostic approach
The golden standard for the diagnosis of PVOD is histological examination of lung tissue which requires surgical lung biopsy. However, in patients with pulmonary vasculopathy, which is present in the majority of SSc patients, the mortality risk of this procedure is significant, making this procedure undesirable. Nowadays, a combination of HRCT, arterial blood sampling, pulmonary function testing, bronchoalveolar lavage (occult alveolar haemorrhage) and right heart catheterization can be used in the diagnostic procedure. PVOD is suspected when HRCT of the thorax shows the triad of lymph node enlargement, centrilobular ground-glass opacities and thickening of septal lines in SSc patients with precapillary PH.8,9 The presence of PAH is confirmed with catheterization when the mean PAP at rest exceeds 25 mmHg. In PVOD, measured values of PCWP are in the normal range (<15 mmHg). PCWP reflects pressures in the larger veins, which are typically less affected in PVOD. The diagnosis of PVOD is suggested by presence of pre-capillary PH, low DLCO in pulmonary function test and HRCT abnormalities as described. Differentiation between PVOD and PAH remains challenging. The specific alterations on HRCT are common in PVOD; presence of the triad has a sensitivity of 75% and a specificity 85%. 5 In our case, there was clear dyspnea on exertion with hypoxaemia and decreased DLCO which could not be explained by ILD. The CT’s showed dilatation of the pulmonary artery, ground glass opacifications and minimal septal thickening. Given the clear clinical hypoxia and the rapid clinical deterioration in combination with HRCT and catheterization findings, the challenging diagnosis of PVOD was preferred.
Treatment options
Treatment options for PVOD are limited; data supporting vasodilator therapy are restricted to case-reports and case-series. Besides, studies using vasodilators show conflicting results. Starting vasodilators carries a risk as possibly the treatment results in dilatation of the arterioles, and not of the fibrotically changed venous vessels which may precipitate pulmonary edema. Epoprostenol (prostacycline analogue; vasodilator and thrombocyte aggregation inhibitor) as a bridge to transplantation has been used in some cases. In our patient, treatment was started with oral double therapy with ambrisentan (endothelin-receptor-antagonist) and tadalafil (phosphodiesterase type-5 (PDE5) inhibitor) under supervision of specialized nurses since benefit from upfront combination therapy in patients with SSc has been shown.10 Also furosemide, spironolactone and oxygen suppletion were started. Efficacy and safety assessments were performed routinely which included clinical response, adverse events, laboratory assessments and 6-MWT. Currently, our patients are not eligible for lung transplantation, due to her morbid obesity. Up till now the medication shows reasonable clinical efficacy. The only complication so far was moderate increase of liver enzymes, without signs of liver dysfunction (Gamma GT 158 U/L, alkali phosphatase 133 U/L, alanine aminotransferase (ALAT) 30 U/L, aspartate aminotransferase (ASAT) hemolytic). To assess the efficacy of treatment, right heart catheterization and 6-MWT were repeated after 5 months. The mPAP decreased from 39 to 30 mmHg, PVR decreased from 3.3 to 1.7 Wood Units, CO increased from 8 to 9.9 L/min and the 6-MWT improved from 123 to 405 meter. Given morbid obesity, the patient will be referred for gastric bypass in the near future, as in the end, and lung transplantation remains the only alternative for the majority of patients with PVOD.
Conclusion
We here demonstrate a case of a 45-year-old female, with a recent diagnosis of limited SSc that presents with rapidly progressive dyspnea and hypoxia on exertion. Based on the combination of rapid clinical deterioration in combination with typical findings on HRCT and increased pulmonary arterial pressure with normal wedge pressure on RHC, the challenging diagnosis of PVOD was made. Strikingly, echocardiography and laboratory tests did not show any abnormalities in favour of PH. With this case, we aim to emphasize that PVOD and PAH are both conditions within one large spectrum of diseases. Also in SSc, clinical distinction between PAH and PVOD is difficult but important to offer appropriate therapy and clinical monitoring, as patients with PVOD have a worse prognosis, frequently show rapid deterioration and are less prone to respond to medication.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Informed consent
Signed informed consent was obtained for patient information and images to be published.