
Abstract
Rationale:
Decompressive craniectomy (DC) is beneficial in people with malignant middle cerebral artery infarction. Whether DC improves outcome in spontaneous intracerebral haemorrhage (ICH) is unknown.
Aim:
To determine whether DC without haematoma evacuation plus best medical treatment (BMT) in people with ICH decreases the risk of death or dependence at 6 months compared to BMT alone.
Methods and design:
SWITCH is an international, multicentre, randomised (1:1), two-arm, open-label, assessor-blinded trial. Key inclusion criteria are age ⩽75 years, stroke due to basal ganglia or thalamic ICH that may extend into cerebral lobes, ventricles or subarachnoid space, Glasgow coma scale of 8–13, NIHSS score of 10–30 and ICH volume of 30–100 mL. Randomisation must be performed <66 h after onset and DC <6 h after randomisation. Both groups will receive BMT. Participants randomised to the treatment group will receive DC of at least 12 cm in diameter according to institutional standards.
Sample size:
A sample of 300 participants randomised 1:1 to DC plus BMT versus BMT alone provides over 85% power at a two-sided alpha-level of 0.05 to detect a relative risk reduction of 33% using a chi-squared test.
Outcomes:
The primary outcome is the composite of death or dependence, defined as modified Rankin scale score 5–6 at 6 months. Secondary outcomes include death, functional status, quality of life and complications at 180 days and 12 months.
Discussion:
SWITCH will inform physicians about the outcomes of DC plus BMT in people with spontaneous deep ICH, compared to BMT alone.
Trial registration:
ClinicalTrials.gov Identifier: NCT02258919

Introduction and rationale
Non-traumatic intracerebral haemorrhage (ICH) accounts for 3.41 million (2.97–3.91) of about 12.2 million strokes worldwide each year. 1 ICH is the most serious and least treatable form of stroke. How to effectively treat people with severe deep supratentorial ICH is a major unresolved issue in acute stroke management. Apart from a care bundle protocol for intensive blood pressure lowering and physiological control management, including immediate anticoagulation reversal, most pharmacological and surgical treatment approaches have failed to reduce mortality and morbidity in people with ICH.2–4 Neither the ‘International Surgical Trials in Intracerebral Haemorrhage’ (STICH and STICH II) nor the ‘Minimally Invasive Surgery with Thrombolysis in Intracerebral haemorrhage Evacuation’ (MISTIE) trials showed superiority of surgical treatment approaches compared to best medical treatment (BMT) in people with ICH.5–7
Decompressive craniectomy (DC) within 48 h of onset in people with malignant middle cerebral artery infarction reduces mortality and improves functional outcome, with a number needed to treat to save one patient’s life of two.8,9 DC is also a standard procedure in people with ICH due to sinus venous thrombosis, herpes encephalitis or ruptured intracranial aneurysms. However, whether DC is beneficial in people with ICH is unknown. DC may relieve the mass effect, decrease perihaematomal tissue pressure, improve blood flow, reduce secondary brain damage and improve outcome. Two studies in animals with experimental ICH have shown that DC reduces mortality and improves outcome compared to non-surgical treatment.10,11 A retrospective case–control study in humans has shown that DC is safe and feasible and may reduce mortality when compared to matched controls receiving BMT. 12 However, DC is a major surgical intervention carrying considerable risk for haemorrhagic, infectious and cerebrospinal fluid (CSF) disturbance-related complications. 13
Early minimally invasive surgery (MIS) for ICH removal can remove large parts of the clot, is potentially less prone to complications, and therefore offers an alternative to major surgery. 14 Several RCTs are comparing MIS plus BMT with BMT alone: MIND (NCT03342664), investigating the Artemis™ device within 72 h has been halted; EVACUATE (NCT04434807), assessing the Aurora Surgiscope® within 8 h, DIST (NCT05460793), investigating MIS within 8h and EMINENT-ICH (NCT05681988) investigating MIS within 24 h are ongoing and results are not expected before 2026/2027. ENRICH-ICH (NCT02880878), testing the Brainpath® device within 24 h showed a beneficial effect for lobar, but not for deep supratentorial ICH. MIS seems to be very promising in people with lobar ICH, but the beneficial effect for deep ICH has yet to be proven.
SWITCH complements these trials by investigating DC in people with deep supratentorial ICH. It will inform physicians whether DC plus BMT in people with spontaneous deep supratentorial ICH will improve outcome at 6 months compared to BMT alone. SWITCH has the potential to answer a major clinical dilemma for stroke physicians and to prompt changes in guidelines to benefit patients.
Methods
Study design
SWITCH is a multicentre, randomised-controlled (1:1), two-arm trial comparing DC and BMT with BMT alone, following spontaneous, supratentorial ICH. Participants’ flow is depicted in Figure 1. The trial is conducted in 42 stroke centres in Switzerland, Austria, Belgium, Finland, France, Germany, Netherlands, Spain and Sweden. The first participant was enrolled in October 2014.
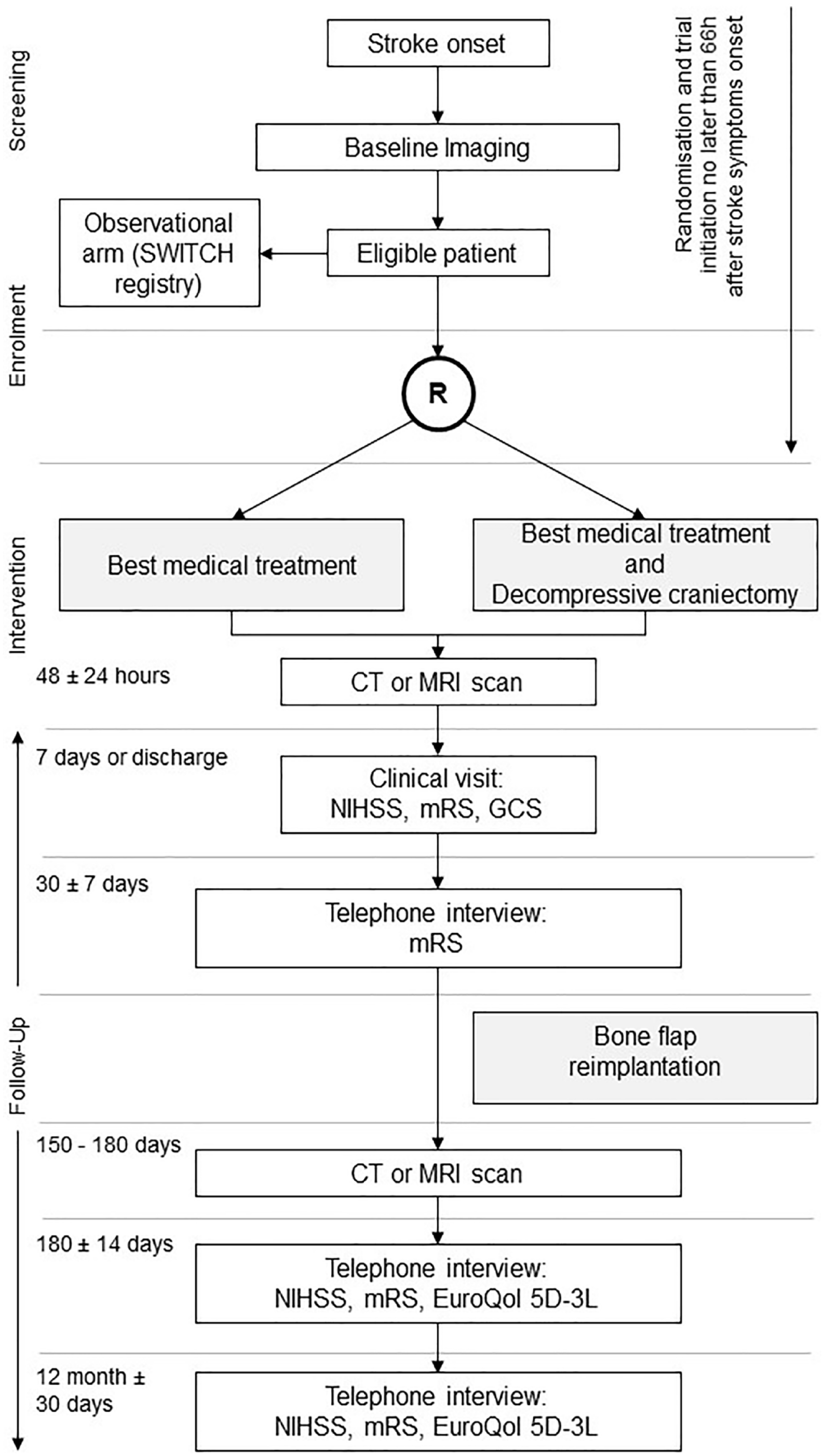
Trial flowchart.
Population
The trial population consists of adults (⩽75 years) presenting with a life-threating, supratentorial, deep (i.e. basal ganglia or thalamus that may extend into cerebral lobes, ventricles, or subarachnoid space) ICH. Inclusion and exclusion criteria are listed in Table 1.
Inclusion and exclusion criteria.

ICH: intracerebral haemorrhage; GCS: Glasgow coma scale; NIHSS: National Institutes of Health Stroke Scale; INR: international normalised ratio; mRS: modified Rankin Scale.
Randomisation and blinding
Randomisation is performed within 66 h after symptom onset; DC no later than 6 h after randomisation. Participants are randomly assigned to one of the two treatment arms using probabilistic minimisation. Allocation is stratified by NIHSS (⩽20 vs
Treatment
After clinical evaluation, all potential participants undergo either computed tomography (CT) or magnetic resonance imaging (MRI) to diagnose the ICH and define stable haematoma volume. All enrolled patients receive BMT.15,16 Participants randomised to the experimental group receive DC (diameter ⩾12 cm) without haematoma evacuation.17,18 The bone flap is reinserted within 1–5 months after DC, followed by a postoperative CT scan.
Clinical and imaging evaluation
Clinical and radiological examination with CT or MRI are performed 48 ± 24 h after enrolment. Clinical follow-up examination is at 7 days or discharge. At 30 ± 7 days, the modified Rankin scale (mRS) score is assessed during a clinical examination or telephone interview. Bone flap reinsertion is performed at 30–150 days if indicated. At 150–180 days a CT or MRI is performed. At 180 ± 14 days and at 12 months ±30 days, the mRS is assessed by an independent, blinded certified rater using validated standards during either a clinical visit or telephone interview. The EuroQol is assessed at 6 and 12 months.
Primary outcome
The primary outcome is a composite of death or dependency (mRS 5 or 6) at 6 months.
Secondary outcomes
The secondary clinical efficacy outcomes are: (1) composite of death or dependency at 30 days and 12 months; (2) death at 7, 30 and 180 days and at 12 months; (3) mRS score of 0–3 versus 4–6 at 30 and 180 days, and at 12 months; (4) categorical shift in mRS score at 180 days and at 12 months; (5) quality of life, measured with the EuroQol at 180 days and at 12 months; (6) NIHSS score at 7 and 180 days; (7) Glasgow coma scale (GCS) at 7 and 180 days; (8) length of hospital stay; (9) requirement for permanent residential care at 180 days and at 12 months. The secondary efficacy outcomes evaluated on imaging are: (1) extent of infarction/brain damage on CT/MRI at 180 days; (2) midline shift at 48 h and 180 days; (3) ICH volume at 48 h; (4) haematoma enlargement at 48 h; (5) number of participants with an extracranial ventricular drain at 7 and 180 days; (6) number of participants with a CSF shunt at 180 days; (7) extension of intraventricular bleeding using the Graeb score at 48 h and 180 days 19 (8) surgical removal of haematoma at 7 and 180 days; (9) requirement for minimally invasive procedure (clot lysis, endoscopic procedure) at 7 and 180 days. Safety outcomes are: (1) all serious adverse events (SAE) up to the study end; (2) solicited adverse events (AEs) at 7 and 180 days.
Data safety monitoring board (DSMB)
An independent DSMB meets after 100, 150, 200 and 250 participants have been enrolled and reviews trial conduct and safety data and formally assesses efficacy and futility.
Hypothesis and statistical analysis
The primary statistical null hypothesis that there is no effect of DC plus BMT compared to BMT alone in people with acute, spontaneous, supratentorial ICH is tested against a two-sided alternative.
Sample size calculation
Three hundred participants were planned to be included in SWITCH. As shown in our feasibility study, the proportion of people with unfavourable outcome (mRS 5–6) was 0.53 in the control group. 12 A total sample size of 300 participants provides over 85% power at a two-sided α level of 0.05 to detect a relative risk reduction of 33% using a chi-squared test. This risk reduction corresponds to a risk for an unfavourable outcome of 0.36 in the treatment group.
Interim and statistical analysis
An interim analysis is conducted after 100, 150, 200 and 250 participants have reached data maturity for the primary endpoint. The null hypothesis will be rejected at a two-sided α level of 0.001 at all planned interim analyses. Because of the low α level, the final analysis will not be adjusted and a conventional two-sided α of 0.05 will be used. The trial may also be stopped for futility if the conditional power at an interim analysis is low according to DSMB judgement. Stopping for futility is conservative concerning the type I error rate but as stopping for futility is non-binding, an α adjustment is not done.
The primary endpoint and binary secondary endpoints will be analysed using a Cochran-Mantel-Haenszel chi-squared test; treatment effects will be reported as Mantel-Haenszel risk ratio and risk difference with 95% confidence intervals (CI), all stratified for the factors used at randomisation (except for centre due to the low number of participants in some centres, which may affect the stability of the estimates). The categorical shift in mRS will be analysed using proportional odds logistic regression adjusted for the stratification factors (except for centre) and the Wilcoxon-Mann-Whitney test. Time to death will be presented using a Kaplan–Meier curve and analysed using a stratified log-rank test. The difference in mortality at different time points and in the restricted mean survival time at 12 months will be calculated with 95% CI from flexible parametric survival models adjusted for the stratification factors (except for centre). Continuous endpoints will be analysed using linear regression adjusted for baseline values (if applicable) and stratification factors (except for centre). Length of hospital stay will be analysed using a flexible parametric accelerated failure time model adjusted for the stratification factors (except for centre) and the effect presented as time ratio with 95% CI. CI reflect uncertainty of the estimates and were not adjusted for multiplicity; therefore, they should not be interpreted as hypothesis tests.
The primary analyses will be performed according to the intention-to-treat principle including all randomised participants in the group to which they were allocated regardless of crossovers or other protocol deviations. Missing data will be imputed with multiple imputation methods if there are more than 5% missings. For outcomes that are not defined in case of death, that is, all outcomes except death itself and the mRS-based outcomes, we will calculate separate estimands for (1) participants alive and (2) all participants (details provided in the statistical analysis plan). A secondary per-protocol analysis will exclude people who violated eligibility criteria, did not receive treatment as assigned or were assessed outside the visit windows. A complete case approach will be used in case of less than 10% crossovers. Otherwise, we will use inverse probability of censoring weighting to emulate an unbiased scenario.
The primary outcome will be analysed for subgroups defined by the stratification factors (except centre), ICH volume, presence of intraventricular haemorrhage, blood pressure at randomisation, use of aspirin or other anti-platelet medication, baseline midline shift, presence of spot sign, and time from symptom onset to randomisation. We will use logistic regression models with the treatment group, the subgroup and their interaction as covariates.
Safety endpoints will be analysed in all randomised participants who received one of the study interventions according to the treatment they actually received. AEs and solicited AEs will be summarised by treatment group and compared using risk differences with 95% CI and Fisher’s exact test. The incidence of SAEs and the number of participants with at least one SAE will be shown by treatment group for: (1) all SAEs and (2) SAEs that are probably or definitely related to the intervention according to investigator judgement. They will be compared between groups using a risk difference and Fisher’s exact test, and an incidence rate ratio and an exact Poisson test, respectively.
Study organisation and funding
SWITCH is an investigator-initiated clinical trial sponsored by the University Hospital Bern. The trial is financially supported by the Swiss National Science Foundation (SNSF 150009), the Swiss Heart Foundation (SHF), the Inselspital Stiftung, and a small unrestricted grant from Boehringer Ingelheim. SWITCH is managed by the Neuro Clinical Trial Unit at the Neurocenter, University Hospital Bern. The electronic clinical trial management system containing the case report form, central data monitoring and statistical analyses is provided by CTU Bern at the University of Bern.
Changes to the protocol and statistical analysis plan during conduct of the trial
The protocol was amended five times (one major and four minor amendments) and the statistical analysis plan once over the course of the trial. The appendix provides a summary of all changes. Major changes concerned selected secondary outcomes and relate to explicit specifications of timepoints which were missing for some (e.g. GCS or imaging-based endpoints), deletion of some (e.g. dependency defined as an mRS of 5 because this cannot be interpreted easily and the full mRS and two dichtomized versions are already included, and time to randomisation as this is not an endpoint but rather a baseline variable), and addition of some surgical endpoints that were mentioned in the statistical analysis plan but not in the protocol. The main changes to the statistical analysis plan relate to technical specifications (e.g. exact method for calculating CI for risk difference), change in the effect measure for the analysis of length of stay (change from restricted mean survival time difference to time ratio calculated from an accelerated failure time model), dropping of two subgroups, and more detailed specification of the approach for analysis of endpoints that are not defined in case of death.
Trial status
Recruitment of participants in SWITCH was stopped by the steering committee on 30/04/2023 due to lack of funding after randomisation of 201 participants.
Discussion
SWITCH aims to test whether DC without haematoma evacuation plus BMT versus BMT alone improves outcome in people with severe deep supratentorial ICH. SWITCH is a proof-of-concept trial with narrow inclusion and exclusion criteria. It focuses on a selected ICH population with deep supratentorial ICH who are severely affected but not comatose and excludes people with lobar haemorrhages, who have been included in most other surgical ICH trials (i.e. MISTIE, STICH, ENRICH-ICH, etc.). Furthermore, SWITCH has selected centres with an established expertise in DC, willing to randomise people with severe ICH into a trial. Narrow selection criteria always hamper recruitment rate, especially in academic investigator-initiated clinical trials with a limited budget. Although the sample size appears to be low and the recruitment rate slow, SWITCH is completely in line with previous trials. SWITCH randomised 201 participants over a period of 8.6 years (recruitment rate 23.4 people/year), HAMLET 20 randomised 64 people within 5 years (12.8 people/year), DESTINY 21 32 people in 4 years (8 people/year), DECIMAL 22 38 people in 4 years (9.5 people/year) and DESTINY II 23 112 people within 3 years (37.3 people/year), even though the latter had broad inclusion and exclusion criteria. SWITCH will therefore become the largest trial on DC ever to be performed in the field of stroke. The long recruitment period reflects the challenge of running investigator-initiated surgical trials in highly selected people with ICH and is a testament to the determination of the investigators to answer an important clinical question in ICH research despite limited resources.
The primary endpoint is the composite of death or dependency at 6 months, measured with the mRS. Secondary outcomes are mortality, dependency and quality of life. For secondary outcomes, exploratory analyses will be performed and the estimates, with 95% CI, will be reported without any formal test or any adjustment for multiple testing. SWITCH is the only trial so far on DC in highly selected people with ICH and therefore secondary exploratory analyses are necessary for formulating hypotheses for future randomised-controlled trials.
The SWITCH trial has several limitations. First, it was performed in a selected population with severe supratentorial ICH and will not answer the question whether people with lobar or infratentorial ICH will benefit from DC. Second, due to the long recruitment period of almost 10 years, several minor protocol revisions were performed. None of the changes had an impact on the primary outcome or on key secondary outcomes. For the sake of transparency, all changes made can be seen in the protocol revision history and all protocol versions are included in the appendix. Third, SWITCH did not reach the target sample size and was stopped by the steering committee due to lack of funding. The decision of the steering committee to stop the trial prematurely and any changes to the protocol were performed without inspection of the data or access to trial results.
SWITCH will inform physicians whether DC plus BMT in people with spontaneous deep supratentorial ICH will improve outcome at 6 months compared to BMT alone. SWITCH has the potential to answer a major clinical dilemma for stroke physicians and to prompt changes to future guidelines that may benefit patients.
Supplemental Material
sj-pdf-1-eso-10.1177_23969873241231047 – Supplemental material for Swiss trial of decompressive craniectomy versus best medical treatment of spontaneous supratentorial intracerebral haemorrhage (SWITCH): an international, multicentre, randomised-controlled, two-arm, assessor-blinded trial
Supplemental material, sj-pdf-1-eso-10.1177_23969873241231047 for Swiss trial of decompressive craniectomy versus best medical treatment of spontaneous supratentorial intracerebral haemorrhage (SWITCH): an international, multicentre, randomised-controlled, two-arm, assessor-blinded trial by Urs Fischer, Christian Fung, Seraina Beyeler, Lukas Bütikofer, Werner Z’Graggen, Florian Ringel, Jan Gralla, Karl Schaller, Nikolaus Plesnila, Daniel Strbian, Marcel Arnold, Werner Hacke, Peter Jüni, Alexander David Mendelow, Christian Stapf, Rustam Al-Shahi Salman, Jenny Bressan, Stefanie Lerch, Claudio L. A. Bassetti, Heinrich P. Mattle, Andreas Raabe and Jürgen Beck in European Stroke Journal
Footnotes
Acknowledgements
Editorial assistance was provided by Susan Kaplan. We thank Sven Trelle for methodological support and Mattia Branca for supporting the trial statistician.
Author contributions
Design: UF, CF, WZ, FR, JG, KS, NP, DS, MA, WH, PJ, DAM, CS, JBr, SL, CLAB, AR, JB. Revision: SB, LB, RAAS. Draft: UF, CF, LB, SB and JB. All authors have revised the manuscript and made critical comments.
Declaration of conflicting interests
This is an academic investigator-initiated trial. No conflicts of interest related to this manuscript need to be declared.
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This trial is supported by the Swiss National Science Foundation (SNSF 150009), the Swiss Heart Foundation, the Inselspital Stiftung and a small unrestricted grant from Boehringer Ingelheim.
Informed consent
Written informed consent according to country-specific requirements is an inclusion criterion for the study.
Ethical approval
Ethical approval was granted by the Cantonal Ethics Commission (KEK) Bern Switzerland (REC number: 163/14, PB_2016-02332), and subsequently by all local authorities and/or, if applicable, by national lead ethics committees at all participating sites.
Guarantor
Professor Urs Fischer
Contributorship
Design: UF, CF, WZ, FR, JG, KS, NP, DS, MA, WH, PJ, DAM, CS, JBr, SL, CLAB, AR, JB. Revision: SB, LB, RAAS. Draft: UF, CF, LB, SB and JB. All authors have revised the manuscript and made critical comments.
ORCID iDs
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.