
Abstract
Background:
Current evidence suggest that 25%–33% of stroke-survivors develop post-stroke cognitive impairment (PSCI). The licensed drug Maraviroc, a CCR5-antagonist, is postulated to act via a neuroprotective mechanism that may offer the potential of preventing progression to vascular dementia. Our hypothesis: Maraviroc may have the potential to augment learning skills and cognitive performance by affecting synaptic plasticity, along with neuro-inflammatory modulation in patients with cerebral small vessel disease (SVD) and PSCI.
Design:
MARCH is a multi-center, double-blind randomized-control Phase-II trial of Maraviroc 150 or 600 mg/day versus placebo for 12-months in five stroke centers in Israel. Included are patients diagnosed with recent (1–24 months) subcortical stroke who experience mild PSCI and have evidence of white matter lesions and SVD on neuroimaging.
Outcomes:
Primary outcomes: 1. Change in cognitive scores. 2. Drug related adverse events. Secondary outcomes: change in functional and affective scores, MRI-derived measures, inflammatory markers, carotid atherosclerosis, cerebrospinal-fluid biomarkers in a sub-study. A sample size of 60 in each treatment group and 30 in the placebo group (total – 150 participants) provides 80% power between the treatment and the placebo groups.
Conclusions:
The results of this work could lead to a novel, readily available, therapeutic avenue to reduce PSCI, and possibly other pathologies. This study will test safety and effectiveness of Maraviroc in limiting cognitive deterioration and/or post stroke cognitive impairment in patients with cerebral small vessel disease.
Schedule:
First-patient first-visit was May 2021. Recruitment to complete in 2023, follow-up to complete in 2024.
Introduction and rationale
Stroke survivors are at risk for developing cognitive changes and hasten Alzheimer’s disease pathology.1,2 White matter lesions (WML) are major contributors to post-stroke cognitive impairment (PSCI) and may indicate small vessel disease (SVD), demyelination, or inflammatory processes. 3 Currently there are no optional therapies for PSCI.
Several very recent preclinical experiments and observational studies in stroke survivors suggest that the commercially available medication Maraviroc, a CCR5 antagonist approved for use in Human Immunodeficiency Virus (HIV)-1 patients, may augment learning skills and cognitive performance.
CCR5 is a pro-inflammatory receptor expressed in microglia, astrocytes and neurons in many brain regions, and is uniquely expressed in cortical neurons after stroke. 4 This receptor has been reported to be involved in learning and memory. 5 Ligand binding to CCR5 is known to modulate several parallel signaling cascades implicated in long-term potentiation, hippocampus-dependent memory, and neocortical experience-dependent plasticity,4,5 including the suppression of adenyl cyclase, as well as the activation of the PI3K/AKT and MAPK/CREB signaling. These findings support the application of brain permeable CCR5 antagonists, not only as a combination drug in antiretroviral therapy, but also as a treatment for cognitive deficits caused by HIV. Indeed, Maraviroc has already been reported to improve cognitive function in chronic HIV-infected patients experiencing HIV-associated dementia (HAD).6–8 In individuals suffering from HIV/AIDS treatment with Maraviroc (150 or 300 or 600 mg vs placebo) showed a large effect on cognitive function observed after 6 and 12 months (d = 0.77).7,8
Our group, along with other collaborators, have recently tested the effects of the naturally-occurring CCR5-Δ32 loss of function mutation in a large cohort of mild stroke patients. In this exclusive prospective cohort of 575 first-ever stroke patients (TABASCO cohort), 9 free of dementia at baseline, who were followed for several years to assess for PSCI; about 15% were carriers of the mutation. This group showed significantly better cognitive and functional outcomes 2 years post-stroke.
There are a two proposed mechanisms of action for Maraviroc in preventing PSCI: (1) neuromodulation and improved synaptic plasticity and (2) local immunomodulatory effect that reduces ischemia related inflammatory reaction and chronic inflammation. Immuno-histochemical analysis of human brain tissue of AD patients, revealed the expression of CCR5 in the cortex and hippocampus. 10 A recent report demonstrated that administration of low-dose Maraviroc attenuated glial cells activation, maintained endothelial monolayer integrity, reduced T cells infiltration, and neuroinflammation in hemiparkinsonian monkeys. 11 Additionally, recent human data suggests an anti-atherogenic effect: Maraviroc led to significant improvements in endothelial function and carotid atherosclerosis, 12 signs that usually accompany Vascular Dementia (VaD). Moreover, it was found to decrease inflammation and the number of membrane adhesion molecules, which are also upregulated in the cerebral microvasculature in AD patients.
Maraviroc is a small molecule, orally-administered, metabolized by CYP3A4, with a good pharmacokinetic profile, relatively low protein binding and high bioavailability. The drug has been in the market since 2007 and has a known and satisfactory safety profile. Main adverse events include upper respiratory tract infections, cough, pyrexia, and rash.
We hypothesize that Maraviroc may be a safe and effective treatment for preventing cognitive deterioration in recent subcortical stroke patients experiencing PSCI, with WML and SVD. Consistent blocking of CCR5 by Maraviroc may protect both blood vessels and neurons, thus prevent the deleterious consequences of another stroke as well as minimizing the deficits of the index event.
Therefore, the Maraviroc in post-stroke patients with Cognitive impairment and white matter Hyperintensities (MARCH) will test Maraviroc in subcortical-stroke patients experiencing PSCI and WML, to assess its safety, and efficacy on halting cognitive and functional progression. As surrogate markers for VaD we will assess its efficacy on SVD lesion progression’ microstructural tissue integrity, brain atrophy using magnetic resonance imaging (MRI)-derived advanced measurements, recurrent vascular events, blood inflammatory markers, and carotid atherosclerosis.
Methods
Design
Multicenter, prospective, randomized, placebo-controlled Phase-II trial conducted in five Israeli stroke centers (Tel Aviv (TASMC), two in Jerusalem (Hadassah and Shaare Zedek), Beer-Sheva (Soroka), Haifa (Rambam)). This choice of centers ensures a diverse ethnic population. Participants are randomized in a 2:2:1 ratio into three groups: low dose Maraviroc (150 mg/day), high dose Maraviroc (600 mg/day), and placebo for a 12 months treatment period. Both treatment groups begin with 2 weeks of low-dose Maraviroc with a dose escalation protocol, for reducing potential adverse effects.
Patient population
Inclusion criteria:
Subcortical symptomatic ischemic stroke at least 1 month and up to 24 months prior to enrollment, compatible with a clinical subcortical stroke syndrome, with neuroimaging showing a symptomatic subcortical infarct.
Age 50 to 86 years.
Fulfill diagnostic criteria for Mild Cognitive Impairment (MCI), which developed after the documented stroke, and indicated by: (i) concern about a change in cognition, in comparison with the person’s previous level; (ii) Montreal Cognitive Assessment (MoCA) score ⩽26; (iii) general preservation of independence in functional abilities.
Neuroimaging showing: (i) periventricular and deep WMLs (grading scale ⩾1 on the Fazekas score); (ii) at least one lacunar infarct; and (iii) absence of large cortical infarcts.
High functional status as assessed by a modified Rankin Score (mRS) ⩽3.
Willingness and ability to complete a 3-tesla MRI scan.
Exclusion criteria:
Diagnosis of dementia or significant cognitive impairment or a MoCA score <17, or other neurological conditions that affects cognition and mobility (multiple sclerosis, Parkinson’s disease, epilepsy, Huntington’s disease, amyotrophic lateral sclerosis, active seizure disorder, attention deficit disorder).
Hemorrhages and cerebral edema (e.g. subarachnoid hemorrhage, intracerebral hemorrhage, subdural hematoma, epidural hematoma. Microbleeds are allowed).
Patients in a state of coma or with severe disturbance of consciousness, aphasia, agnosia, or deafness that subsequently affects expression and communication.
Significant systemic medical illness including drug abuse, severely disturbed liver (confirmed and unexplained impaired hepatic function as indicated by screening AST or ALT ⩾3 the upper limit of normal (ULN) or total bilirubin ⩾2 ULN), kidney or lung function (Chronic kidney disease of stage ⩾4, according to the National Kidney Foundation Kidney Disease Outcomes Quality Initiative guidelines for chronic kidney disease), anemia (Hemoglobin <10), hypothyroidism, or uncontrolled diabetes.
Presence of cortical involvement on neurologic examination including aphasia, hemineglect, etc.
Diagnosis of a genetic cause of VaD (e.g. CADASIL).
Concurrent drug therapy that may affect cognitive function: cholinesterase inhibitors, cannabinoids.
Inability to meet the specific scanning requirements of the 3T MRI.
History of hepatitis or elevated hepatic transaminases or bilirubin; positive serology for Hepatitis B or C; positive serology for HIV.
Serum creatinine over 1.8 mg/dL.
Current or past diagnosis of significant psychiatric comorbidity, and substance/alcohol use disorders other than nicotine in the past year. Subjects with a history of stroke-related depression or anxiety can be included.
Suicidal behavior within the past year.
Diagnosis of attention deficit disorder.
Prolongation of the corrected QT interval.
Use of drugs with possible interactions with Maraviroc.
Known allergies, hypersensitivity, intolerance, or contraindication to Maraviroc or its excipients.
Subject has received an investigational drug, or used an invasive investigational medical device within 60 days before the planned first dose of study drug or currently enrolled in an investigational study.
Any condition for which, in the opinion of the investigator, participation would not be in the best interest of the subject.
History of a major surgery within 2 weeks before screening.
Pregnant or breastfeeding women or women of childbearing age not taking contraception.
Protocol approvals, registrations, and patient consents
This study was registered as ClinicalTrials.gov Identifier: NCT04966429. All participants signed a written informed consent forms, approved by the local ethics committee (REC Ref: 0625-20-TLV).
Randomization and blinding
Baseline information for eligible participant is entered to a computer algorithm randomizes participants at a 2:2:1 ratio to a study group. All members of the study team and the subjects are blinded as to allocation. Apart from the study project manager, other staff performing all assessments will not be aware of the treatment allocation. An external sub-contractor will conduct placebo manufacturing and study drug capsulation in study kits and will supply the kits to each site’s pharmacy.
Intervention
Maraviroc or placebo are started within 1–24 months of stroke onset. Both active treatment groups will have two phases of treatment: Phase 1; Run-in phase – all patients in the treatment group will receive low dose Maraviroc (150 mg/day) for 2 weeks to ensure tolerability. Phase 2; active phase-after a 2-week run-in phase period, patients from the treatment group are assigned according to their randomization to receive either low (150 mg/day) or high dose (600 mg/day) therapy for 46 weeks. Thus, a 2-week supply of capsules is handled to participants after consent has been signed, baseline measurements obtained, and randomization assigned. During the run-in phase, all the patients will receive one capsule per day in the morning: both treatment groups will receive Maraviroc 150 mg/day and the placebo will receive one capsule of placebo. The second supply is provided near the end of the second week on medication. Participants will take two capsules BID, either Maraviroc 150 or 600 mg or placebo.
Primary outcome
The difference in mean change from baseline to week 48 between maraviroc 150 mg/day, maraviroc 600 mg/day, and placebo-treated patients in the Toronto Cognitive Assessment (TorCA). 13 The analysis will include all randomized patients, with patients grouped according to the treatment assigned at randomization.
Safety: The proportion of participants suffering from adverse events related to the study drug, as well as recurrent vascular events and, cardiovascular related deaths.
Secondary outcomes
The difference in mean change from baseline to week 48 between maraviroc 150 mg/day, maraviroc 600 mg/day, and placebo-treated patients in the cognitive scores: Key secondary outcome: the Clinical Dementia Rating (CDR) Sum of Boxes. 14
The difference in mean change from baseline to week 48 between maraviroc 150 mg/day, maraviroc 600 mg/day, and placebo-treated patients in the cognitive scores: the Montreal Cognitive Assessment (MoCA) and the global cognitive score based on repeatable computerized battery of cognitive tests (Neurotrax).
The difference in mean change from baseline to week 48 between maraviroc 150 mg/day, maraviroc 600 mg/day, and placebo-treated patients in the functional scores as assessed by: the stroke impact scale (SIS), instrumental activities of daily living (IADL) score, gait and balance scores, and Reintegration to Normal Living Index (RNL).
The difference in mean change from baseline to week 48 between maraviroc150 mg/day, maraviroc 600 mg/day, and placebo-treated patients in the severity of affective symptomatology as assessed by: the Center for Epidemiologic Studies–Depression (CES-D), Geriatric depression scale (GDS), the Generalized anxiety disorder (GAD-7).
The difference in mean change from baseline to week 48 between maraviroc 150 mg/day, maraviroc 600 mg/day, and placebo-treated patients in MRI-derived advanced measures: volumetric changes in whole brain volume, parenchymal brain fraction (PBF), white matter (WM) and gray matter (GM) volumes, and volumes of deep structures related to cognition such as thalami and hippocampi. Additionally, changes in microstructural tissue integrity and connectivity as assessed by DTI analysis, extent of SVD burden.
The difference in mean change from baseline to week 48 between maraviroc 150 mg/day, maraviroc 600 mg/day and placebo-treated patients in the cervicocranial vascular atherosclerotic load as assessed by: sonographic assessment of the cervical Carotid artery and MRI derived measures of intracranial atherosclerosis using high-resolution vessel wall imaging (VWI) and vessel narrowing on MR-angiography.
The difference in mean change from baseline to week 48 between maraviroc 150 mg/day, maraviroc 600 mg/day, and placebo-treated patients in blood biomarkers: inflammatory and endothelial function profile.
In a subgroup recruited in TASMC, change in vasomotor reserve (VMR), assessed using transcranial Doppler.
In a subgroup of consenting study participants – the difference at week 24 between maraviroc 150 mg/day, maraviroc 600 mg/day, and placebo-treated patients in CSF biomarkers: cytokine concentrations, Aβ1-42, t-tau, p-tau 181, and S100β (a biochemical marker of inflammation that indicates astrocyte activation).
Data monitoring committee
An independent data monitoring committee (iDMC) is established; chair Dr. Sivan Bloch (Carmel Medical Center). Other members include Prof. Ronen Ben-Ami (TASMC), Prof. Shlomo Berliner (TASMC), and Dr. Gil Harari (MediStat Ltd). The iDMC will review the data quarterly and will advise on study protocol and safety. After 50% of patients have been enrolled and completed follow-up, the iDMC will perform a pre-specified interim analysis, and will advise on further conduction of the trial.
Assessments and data collection
Baseline and follow-up clinical and cognitive assessments
Patients will undergo a baseline through neuropsychological assessment, using validated tools to asses for cognitive function including: the Clinical Dementia Rating® (CDR®), 13 the Toronto Cognitive Assessment (TorCA), 14 the Montreal Cognitive Assessment (MoCA), and the NeuroTrax computerized cognitive testing (NeuroTrax Corp., Bellaire, TX). 15
These comprehensive neuropsychological evaluations will be repeated on week 24 and week 48 from start of treatment with study drug.
Baseline visit will include: demographic data and socio-economic status; verification of sub-cortical stroke history; medical history; verification of neuroimaging criteria for enrollment; vital signs; physical and neurological exam; electrocardiogram (ECG); functional status: mRS, Instrumental activities of daily living (IADL), stroke impact scale (SIS), Quality of life (QoL), Reintegration to Normal Living (RNL) Index; National Institutes of Health Stroke Scale (NIHSS); detailed cognitive assessment; psychiatric evaluation using the Center for Epidemiologic Studies-Depression (CES-D) scale, Geriatric depression scale (GDS), Generalized anxiety disorder (GAD-7) scale; Gait speed and velocity tests; Carotid Doppler.
Follow up visit on weeks 2, 12, and 36 will include: Physical and neurological exam; vital signs, ECG, mRS, NIHSS, record of Adverse events, Blood tests for monitoring liver and renal function and cytokines.
Follow-up visits on weeks 24 and 48 will include: Physical and neurological exam; vital signs, ECG, mRS, NIHSS, record of Adverse events, QoL, IADL, RNL, Blood tests for monitoring liver and renal function and cytokines, cognitive and psychiatric assessments, gait speed and velocity tests, MRI and Doppler on week 48.
For a sub-group of patients who will consent: cerebrospinal fluid test for cytokines level, Maraviroc concentration and markers of AD pathology (Aβ1-40 and 1-42, t-tau, p-tau).
Study procedures are detailed in Figure 1.
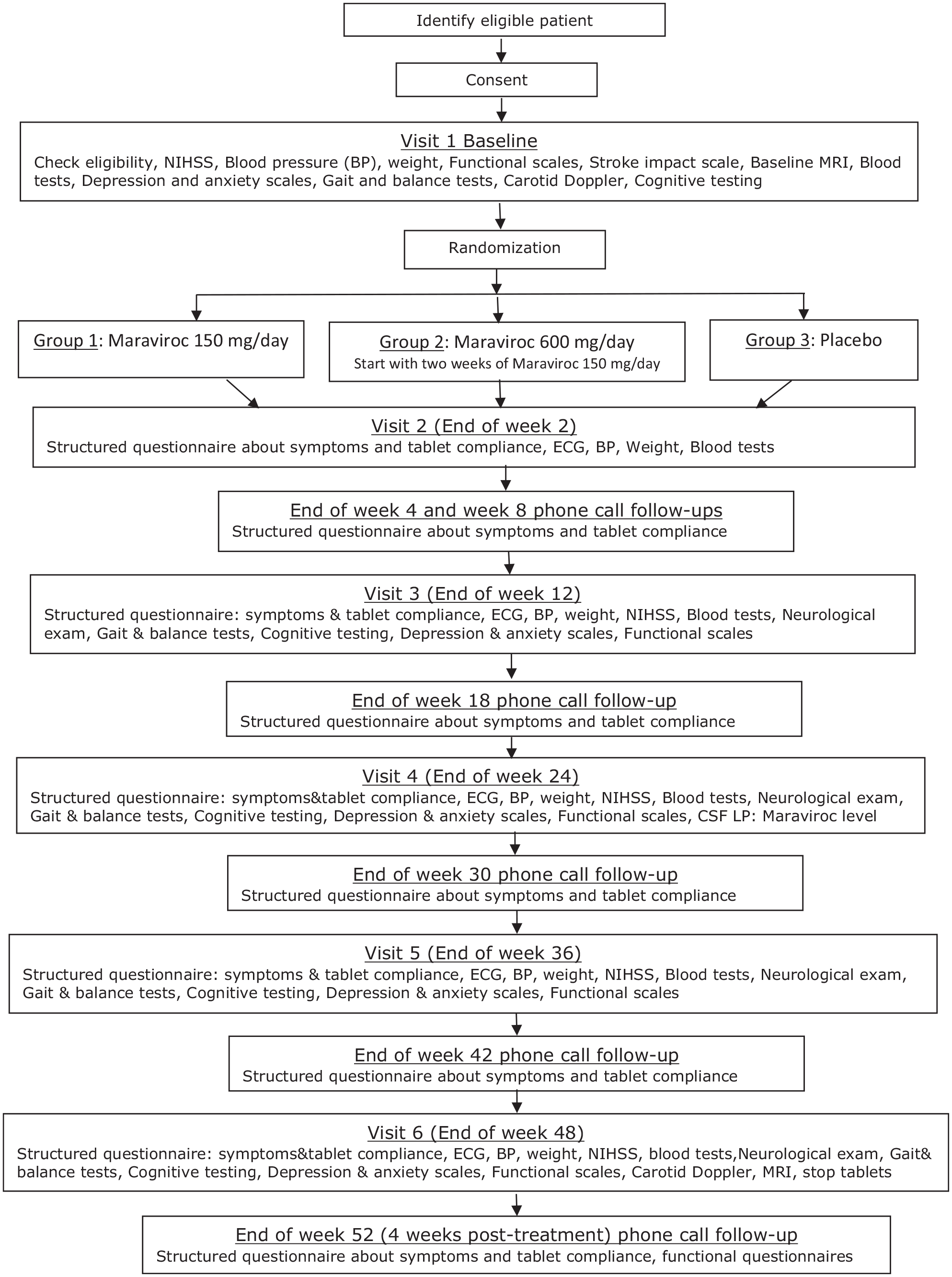
Flow-chart of study procedures.
MRI image acquisition protocol is available in Supplemental Material.
MRI image processing and analysis
MRI acquisition will take place at each site with central analysis at Tel Aviv Sourasky Medical Center (TASMC).
Tissue segmentation and brain atrophy measures: It is performed as previously described.16,17
We will analyze volumetric changes in whole brain volume, parenchymal brain fraction (PBF), white matter (WM) and gray matter (GM) volumes, and volumes of deep structures related to cognition such as thalami and hippocampi. Additionally, changes in microstructural tissue integrity and connectivity as assessed by DTI analysis, extent of SVD burden.
Small vessel disease (SVD) markers: In accordance with STRIVE score. 18 This score determines chronic lacunar infarcts, white matter (WM) hyperintensities (is graded using the Fazekas score), deep and lobar cerebral microbleeds (CMB), and enlarged perivascular spaces (PVS).
DTI analysis of the NAWM: Calculation of the DTI maps is performed using FMRIB Diffusion Toolbox, part of FMRIB software Library (FSL, http://www.fmrib.ox.ac.uk/fsl/). Four different maps are calculated: fractional anisotropy (FA), mean diffusivity (MD), perpendicular diffusivity (λ⊥), and parallel diffusivity (λ||) are calculated.
Intracranial atherosclerotic load assessment: It is available in Supplemental Material.
Ultrasound acquisition and measurements
Carotid artery stenosis is determined by ultrasonographic measurement of the internal carotid arteries. Carotid intima-media thickness (IMT) is measured at the far wall of the common carotid arteries using carotid duplex equipment with a 5–12 MHz linear array transducer and a dedicated software for image acquisition, measurements, and storage (Affiniti 70, Philips).
Transcranial Doppler (TCD, Dolphin 4D, Viasonix, Israel) measurements are performed to assess the velocity of bilateral middle cerebral arteries (MCA) and basilar artery (systolic, diastolic, and mean). Pulsatily index is calculated as a marker for distal cerebral blood flow resistance. Vasomotor reactivity (VMR) is assessed by breath-holding test for 30 s, as previously described. 19 Breath-holding index (BHI) is calculated by an automatic software. Values above of 0.69 are considered as normal response.
Statistical analysis plan
MediStat Ltd, specialized in statistical and data management, will provide statistical support to the trial.
Statistical considerations (sample size rationale)
The planned sample size is 150 participants in the all treatment groups (60 for each dosage) and 30 participants in the placebo group (ratio 2:2:1).
The rationale for sample size calculation was based on demonstrating differences in improvement of cognitive scores.
Based on two interventional trials of Maraviroc in HIV patients with evidence of mild to moderate cognitive impairment7,8 found that impaired subjects showed significant improvement of 0.75 units in global cognitive scores (µ1 for the Maraviroc treatment group), while the placebo group showed improvement of only 0.28 units (µ2) after 6 months.
Sample size justification
A two group t-test with a 0.025 two-sided significance level will have 80% power to detect an Effect size of 0.70 between the treatment group and a placebo group, assuming that the common standard deviation is 0.66, when the sample sizes in the two groups are 60 and 30, respectively. The Effect Size is defined as the expected mean difference between study groups improvement of cognitive scores divided by the standard deviation and based on type 1 error (α) of 0.025 for two-sided test and statistical power of 80%.
A sample size of 60 in each treatment group and 20 in the placebo group is required (ratio 2:2:1), assuming an Effect Size of 0.71. Total of 167 patients is required (assuming drop-out rate of 10%) to ensure at least 150 completed patients.
Statistical analysis methods
All measured variables and derived parameters will be listed individually and will be summary by tabulated and by descriptive statistics.
For categorical variables, summary tables will be provided giving sample size, absolute and relative frequency, and 95% CI (Confidence Interval) for proportions.
For continuous variables, summary tables will be provided giving sample size, arithmetic mean, standard deviation, coefficient of variation (cv%) median, minimum and maximum, and 95% CI (Confidence Interval) for means.
Safety assessment
All safety will be summarized in appropriate tables.
AEs will be coded according to coding dictionaries (MedDRA version 22.0 or higher) and presented in tables by System Organ Class (SOC) and Preferred Term (PT) and by treatment group. Safety will be also assessed by evaluating findings of physical examinations, vital signs, clinical laboratory test results, concomitant medications by treatment group. The changes from baseline in vital signs, clinical laboratory tests results will be displayed.
Efficacy assessments
Primary endpoint: Relative change (%) from baseline in TorCA cognitive score at 12 months
Stage 1: The Paired T-test or Signed rank test for two means (as is appropriate) will be applied for testing the statistical significance of the Relative change (%) from baseline in TorCA cognitive score at 12 months in each treatment group.
Stage 2: The two-sample T-test or Non-parametric Wilcoxon-Mann-Whitney Rank sum test for independent samples (as is appropriate) will be applied for testing the statistical significance of the difference in Relative change (%) from baseline in TorCA cognitive score at 12 months between treatment groups.
Anova model will be applied for testing the difference in Relative change (%) from baseline in TorCA cognitive score at 12 months between treatment groups, adjusted for possible confounders.
To limit Type I error inflation, formal hypothesis testing will proceed down the hierarchy until a test declares p > 0.05, beyond which any subsequent tests will be considered exploratory.
Key secondary endpoint: Relative change (%) from baseline in CDR-SB cognitive score at 12 months.
Stage 1: The Paired T-test or Signed rank test for two means (as is appropriate) will be applied for testing the statistical significance of the Relative change (%) from baseline in CDR-SB cognitive score at 12 months in each treatment group.
Stage 2: The two-sample T-test or Non-parametric Wilcoxon-Mann-Whitney Rank sum test for independent samples (as is appropriate) will be applied for testing the statistical significance of the difference in Relative change (%) from baseline in CDR-SB cognitive score at 12 months between treatment groups.
Anova model will be applied for testing the difference in Relative change (%) from baseline in CDR-SB cognitive score at 12 months between treatment groups, adjusted for possible confounders.
Other secondary endpoints: Relative change (%) from baseline in each other cognitive score at 12 months
Stage 1: The Paired T-test or Signed rank test for two means (as is appropriate) will be applied for testing the statistical significance of the Relative change (%) from baseline in each other cognitive score at 12 months in each treatment group.
Stage 2: The two-sample T-test or Non-parametric Wilcoxon-Mann-Whitney Rank sum test for independent samples (as is appropriate) will be applied for testing the statistical significance of the difference in Relative change (%) from baseline in each other cognitive score at 12 months between treatment groups.
Anova model will be applied for testing the difference in Relative change (%) from baseline in each other cognitive score at 12 months between treatment group, adjusted for possible confounders.
All tests will be two-tailed, and a p-value of 5% or less will be considered statistically significant.
The data will be analyzed using the SAS® version 9.4 (SAS Institute, Cary, North Carolina).
Baseline characteristics for statistical analysis are available in Supplemental Material.
Discussion
MARCH explores the utility of the drug Maraviroc in halting or preventing PSCI, given for 1 year. The main objective is to assess the safety and tolerability of Maraviroc 150 and 600 mg per day versus placebo in patients with subcortical stroke and mild PSCI. The intense follow-up includes thorough record of adverse events, repeated ECG and blood tests for monitoring liver and renal function. At the same time, we will evaluate the efficacy of the two doses versus placebo on cognitive and functional scores, neuroimaging measures, and carotid atherosclerosis. Our primary outcome measure is the TorCA, which is a comprehensive cognitive test. Since this is the first of its kind trial in this population, we added a key secondary outcome (the CDR), that will provide additional insight into the clinical efficacy of Maraviroc. Yet, we consider this to be a pilot or proof of concept study rather than a definitive test of efficacy.
The intense follow-up provides data to inform a future larger, pragmatic trial with less frequent follow-up. The double-blind design will enable clear comparison to matching placebo for either dosage of study drug. While dose escalation of 2 weeks at treatment initiation is used to lessen side effects and enhance compliance, we intend to use a low and high dose in order to minimize the risk of under-dosing and provide a better dose-response data. Due to the known safety profile of Maraviroc, the frequency of significant side effects is expected to be low in both doses.
We believe the length of follow-up and intense clinical, radiological, and biological data collection, will ensure the optimal detection of treatment effect.
The study is conducted in a single country (Israel). However, the geographical locations of the recruiting centers will ensure a wide diversity of the patient population, and enhance generalizability of trial results.
PSCI is a very common sequela of stroke, and presents significant healthcare and quality of life problems for stroke survivors, their families and society. Currently there is no known treatment for PSCI except risk factor control. This study offers a novel treatment and therefore there is no ethical concern with Placebo treatment.
To the best of our knowledge, MARCH appears to be the only ongoing randomized control trial specifically testing agent for prevention of post-stroke dementia and worsening of SVD, with robust endpoints. We hope that MARCH will help inform the future design of trials in lacunar stroke and other clinical presentations of SVD.
Summary and conclusions
This study will provide data on tolerability, safety, and efficacy markers for Maraviroc in subcortical-stroke patients experiencing PSCI, and, if the trial hypothesis is confirmed, MARCH may provide a new strategy for the prevention of post-stroke dementia.
Supplemental Material
sj-docx-1-eso-10.1177_23969873221098857 – Supplemental material for Preventing post-stroke dementia. The MARCH Trial. Protocol and statistical analysis plan of a randomized clinical trial testing the safety and efficacy of Maraviroc in post-stroke cognitive impairment
Supplemental material, sj-docx-1-eso-10.1177_23969873221098857 for Preventing post-stroke dementia. The MARCH Trial. Protocol and statistical analysis plan of a randomized clinical trial testing the safety and efficacy of Maraviroc in post-stroke cognitive impairment by Einor Ben Assayag, Jeremy Molad, Estelle Seyman, Ofer Rotschild, Ehud Zeltzer, Udi Sadeh-Gonik, Noa Bregman, Aviva Alpernas, Yahel Segal, Dafna Ben Bashat, Talya Nathan, Muhamad Hawwari, Oren Tene and Hen Hallevi in European Stroke Journal
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Part the Cloud Gates Partnership & the Alzheimer’s Association [grant number PTCG-20-706182].
Informed consent
Written informed consent was obtained from all subjects before the study. Ethical approval: The study is conducted according to the Declaration of Helsinki and approved by the local ethics committee (REC Ref: 0625-20-TLV).
Guarantor
EBA.
Author Contributions
EBA, HH, and JM conceived the study design. EBA and HH contributed to securing funding. EBA, HH, JM, ES, EZ, NB, DBB, and OT contributed in protocol development. EBA and HH contributed to gaining ethical approval. EBA, HH, JM, ES, OR, EZ, YS, AA, TN, MH, and US contributed to patient recruitment and testing. EBA and HH wrote the first draft of the manuscript. All authors reviewed, edited the manuscript and approved the final version of the manuscript.
Trial registration
ClinicalTrials.gov Identifier: NCT04966429
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.