
Abstract

Case Reports
AS35-139
EFFECTS OF RADIATION THERAPY TO THE NECK IN CHILDHOOD ON THE CAROTID ARTERIES
1University Medical Centre Ljubljana, Neurology, Ljubljana, Slovenia
2Institute of Oncology Ljubljana, Oncology, Ljubljana, Slovenia
Background and Aims
With new treatment options the survival of children with cancer improved over the past decades and with this the risk of developing chronic diseases in this population has also increased. It is known that radiotherapy to the neck increases the risk of carotid disease and stroke in adults but little of that is known in childhood cancer survivors. The aim was to study the local and systemic effects of radiotherapy to the neck in childhood Hodgkin’s disease survivors. In reviewed literature we found few studies which studied possible markers for increased atherosclerosis.
Methods
Our subject is a 48 year old male and was diagnosed with Hodgkin’s disease at 8 years of age. He was treated with radiotherapy to the neck. We performed duplex carotid ultrasonography and FMD. We analysed data of carotid stiffness in survivor. Linear regression was performed to test the relationship between PWV and beta index, elasticity modulus (Ep), augmentation index (AI), arterial compliance (AC).
Results
On duplex carotid ultrasonography several fibrous and calcified plaques were found and without hemodinamically significant carotid stenosis.
We found significant relationship between Ep and PWV (B = 0.021, beta = 0.99; p < 0.01), beta and PWV (B = 0.36, beta = 0.99; p > 0.01), AC and PWV (B = −8.23, beta = 0.925; <0.01) while Al and PWV (B = 0.027; beta = 0.302; p = 0.255) were not related significantly. The FMD value in systole was 4,98%, in diastole 5,31%.
Conclusions
Carotid stiffness in our subject is increased consistently throughout most of the stiffness indices. FMD seems to be decreased. Therefore, endothelial and carotid dysfunction seems to be present in our subject.
Trial registration number
N/A
Clinical Trial Results – Acute Management – Neither Thrombolysis Nor Thrombectomy
AS01-031
THE CLINICAL EFFICACY OF CEREBROLYSIN IN ISCHEMIC STROKE PATIENTS WITH PRE-STROKE MILD COGNITIVE IMPAIRMENT: A PILOT STUDY
1University Clinic, Neurology, Odessa, Ukraine
2Odessa National Medical University, Neurology, Odessa, Ukraine
Background and Aims
Assessment of cognitive improvement after Cerebrolysin® treatment in ischemic stroke patients who suffered from mild cognitive impairment before stroke
Methods
This study had a randomized, placebo-controlled, double-blind design. Patients were randomized within 24 hours post-stroke to either intravenous placebo therapy (n = 19) or Cerebrolysin 40 ml per day (n = 20) for 21 days. Both groups received standard stroke treatment and concomitant treatment for comorbid conditions. Endpoints were MoCA and ARAT scales evaluated three months after discharge. The statistical process was carried out by non-parametric methods with the software Statistica 13.0 (Dell Inc.) based on the LOCF population.
Results
The total number of patients included was 39. The average NIHSS score at time of admission was 9.4 ± 0.2 points. Discharge time was 22.3 ± 0.3 days post stroke. At study endpoint patients in the Cerebrolysin group had improved in the MoCA from 23.8 ± 0.2 at baseline to 25.2 ± 0.1 compared to placebo patients who improved from 23.6 ± 0.2 at baseline to 24.1 ± 0.1, a significant group difference (p <0.001). In the ARAT, Cerebrolysin treated patients improved from 29.9 ± 0.2 at baseline to 39.6 ± 0.4 as compared to placebo patients who improved from 34.2 ± 0.7 to 38.6 ± 0.3 points, a significant group difference (p < 0.05).
Conclusions
Cerebrolysin has a beneficial effect on functional and cognitive performance of mildly cognitive impaired patients who suffered from ischemic stroke. Cerebrolysin was safe and well tolerated
Trial registration number
N/A
Cognition and Vascular Cognitive Impairment
AS26-044
ASSOCIATION BETWEEN FATIGUE AND COGNITIVE FUNCTIONS AT 6 MONTHS IN ISCHEMIC STROKE PATIENTS TREATED WITH ACUTE REVASCULARISATION THERAPY
1CHU Dijon, Neurology, Dijon, France
Background and Aims
Fatigue is among the most prevalent symptoms after stroke, and it is now recognized as an important issue in patients with improved functional recovery thanks to better acute management. Because of limited knowledge on this topic, we aimed to determine the associations between fatigue and cognitive functions at 6 months in ischemic stroke patients.
Methods
Forty six patients with first-ever ischemic stroke treated with acute revascularisation therapy were included in the stroke unit of Dijon, France. At 6 months, we performed clinical examination, evaluation of post-stroke fatigue with the Fatigue Severity Scale (FSS), assessment of anxiety/depression using the HAD scale, and a comprehensive neuropsychological evaluation. Patients with significant fatigue (FFS score > 4) were compared to patients without fatigue.
Results
In our cohort, 35% patients reported significant post-stroke fatigue at 6 months. Patients with fatigue had a greater prevalence of depression, and higher mRs scores. Concerning neuropsychological evaluation, only deficit on divided attention was more frequent in the fatigue group (75% versus 32%, p = 0,032), even in a subgroup of non-depressed patients.
Conclusions
Our study suggests that a deficit on divided attention is more frequent in patients with post-stroke fatigue. Further studies are needed to evaluate whether cognitive rehabilitation strategies may have an impact on fatigue.
Trial registration number
N/A
Rehabilitation – Excluding Clinical Trial Results
AS08-069
CANADIAN PLATFORM FOR TRIALS IN NON-INVASIVE BRAIN STIMULATION (CANSTIM) CONSENSUS RECOMMENDATIONS FOR REPETITIVE TRANSCRANIAL MAGNETIC STIMULATION IN UPPER EXTREMITY MOTOR STROKE REHABILITATION TRIALS
1University of Ottawa, Ottawa Heart Institute, Ottawa, Canada
2University of Toronto, Sunnybrook Research Institute, Toronto, Canada
3Dalhousie University, School of Physiotherapy, Halifax, Canada
4University of British Columbia, Department of Physical Therapy, Department of Physical Therapy, Canada
5Memorial University, Faculty of Medicine, Sr. John’s, Canada
6University of Toronto, Toronto Western Hospital, Toronto, Canada
7University of Calgary, Department of Clinical Neurosciences, Calgary, Canada
8McGill University, School of Physical and Occupational Therapy, Montreal, Canada
9University of Calgary, Departments of Pediatrics and Clinical Neuroscience, Calgary, Canada
10University of Toronto, Rothman Research Institute, Toronto, Canada
11University of Manitoba, Department of Electrical and Computer Engineering, Manitoba, Canada
12McGill University, Department of Kinesiology and Physical Education, Montreal, Canada
13University of Toronto, Department of Psychiatry, Toronto, Canada
14University of Ottawa, Faculty of Health Sciences, Ottawa, Canada
15McGill University, Neurology & Neurosurgery, Montreal, Canada
Background and Aims
Large scale clinical trials demonstrating the efficacy of repetitive transcranial magnetic stimulation (rTMS) for post-stroke functional motor recovery are lacking. Missing consensus regarding the optimal protocol for rTMS in stroke has been a challenge for trial design. It iwas the aim of this consensus process to develop recommendations for the use of rTMS as an adjunct intervention for upper extremity motor recovery in stroke rehabilitation trials.
Methods
The multidisciplinary Consensus Working Group of clinicians and researchers from across Canada identified four consensus themes: 1) patient population; 2) stimulation parameters; 3) rehabilitation interventions; and 4) outcome measures. Comprehensive evidence reviews for each theme were conducted and a weighted dot-voting procedure was used to achieve consensus.
Results
The following consensus recommendations were reached: 1) recruit cortical and subcortical stroke patients between 2 and 12 weeks after stroke who have been identified to need upper extremity rehabilitation 2) randomization to receive 1800 pulses of 1Hz rTMS over contralesional M1 at 120% RMT or sham stimulation 3) followed by 60 minutes of a controlled treatment (GRASP), for a total of 15 sessions. 4) use of a suite of validated core primary outcome measures of impairment, function and ability as well as subjective patient-centered and real-world actigraphy secondary outcomes and MR imaging biomarkers and MEP parameters as exploratory outcomes.
Conclusions
Establishing the CanStim platform and developming these consensus recommendations for an rTMS protocol for clinical trials is a first step toward the translation of non-invasive brain stimulation technologies into the clinic to enhance stroke recovery.
Trial registration number
N/A
Stroke Complications
AS18-070
EARLY RECURRENT EMBOLIC INFARCTION MAY BE AN UNDERRECOGNISED COMPLICATION OF INTRAVENOUS THROMBOLYSIS
1Shenyang First People’s Hospital, Neurology, Shenyang, China
Background and Aims
Early recurrent ischemic stroke (ERIS) can occur during or after intravenous (IV) thrombolysis in patients with tPA. ERIS, as one of the reasons of early neurological deterioration, complicates clinical outcomes by inducing new territorial or large hemispheric infarctions.
Methods
We retrospectively reviewed 518 consecutive AIS patients treated with IV tPA between January 2016 and June 2018. We defined the ERIS as: Firstly, newly emerged identifiable neurological deficits (with an increment of NIHSS ≥ 4-points) during or within 24 hours after intravenous thrombolysis and, secondly ICH excluded and, thirdly new neurological deficit cannot be explained by previously presumed or neuroimaging-shown infarction(s).
Results
Of the 518 patients treated with IV tPA in our center, 5 (0.97%) developed ERIS. Median onset time of ERIS cases were 7 hours after the beginning of IV tPA. All the five patients developed neurological deterioration with median NIHSS of 18 points when recurrence. Compared with patients who did not developed ERIS, ERIS patients were associated with prolonged hospital stay, increased neurological severity (NIHSS), functional outcome (mRS) and in-hospital mortality. Surprisingly, none of the patients was with history of AF or was diagnosed with AF during the hospitalisation. Detailed statistical analysis is underway and will be presented.
Conclusions
Our case series showed ERIS after IV tPA is associated with significantly greater neurological severity and worse functional outcome in ischemic stroke patients, and due to the nature of high prevalence of intracranial stenosis among Chinese population, the results may warrant future observational study from large-scale Asian registry or cohort.
Trial registration number
N/A
Ongoing Trials
WITHDRAWN
AS36-021
MR CLEAN-MED – THE EFFECT OF PERIPROCEDURAL MEDICATION IN PATIENTS UNDERGOING INTRA-ARTERIAL TREATMENT FOR ACUTE ISCHEMIC STROKE: ANTIPLATELET AGENTS, HEPARIN, BOTH OR NEITHER
1Erasmus MC, Radiology & Nuclear Medicine, Rotterdam, The Netherlands
2Erasmus MC, Neurology, Rotterdam, The Netherlands
3Erasmus MC, Public Health, Rotterdam, The Netherlands
4Isala hospital, Neurology, Zwolle, The Netherlands
5Cardiovascular Research Institute Maastricht- Maastricht University Medical Center, Neurology, Maastricht, The Netherlands
6HagaZiekenhuis, Radiology, Den Haag, The Netherlands
7Radboud University Medical Center, Radiology, Nijmegen, The Netherlands
Background and Aims
A considerable proportion of ischemic stroke patients do not recover despite fast and complete recanalization after endovascular therapy (EVT). It is unknown whether periprocedural antithrombotic therapy can improve clinical outcome. The objective of this study is to assess the effect of acetylsalicylic acid (ASA) and unfractionated heparin, alone, or in combination, in patients who undergo EVT.
Methods
MR CLEAN-MED is a multicenter, prospective, randomized, open-label, blinded-endpoint trial using a 2x3 factorial design. We aim to enroll 1500 patients with a clinical diagnosis of acute ischemic stroke and confirmed intracranial anterior circulation occlusion, who will undergo EVT with or without prior intravenous thrombolysis according to standard care. Study interventions: IV treatment with ASA (300 mg), low dose unfractionated heparin (loading dose of 5000 IU followed by 500 IU/hour x 6 hours) and moderate dose unfractionated heparin (loading dose of 5000 IU followed by 1250 IU/hour x 6 hours). Primary outcome is the score on the modified Rankin Scale 90 days after inclusion in the study. Safety endpoints include the occurrence of symptomatic intracerebral hemorrhage.

Results
First patient included on January 22nd, 2018, 100 patients randomized by Jan 23. Seventeen sites participate in the study.
Conclusions
We hypothesize that despite the potentially increased risk of (symptomatic) intracerebral hemorrhage, periprocedural ASA and unfractionated heparin alone or in combination will improve functional outcome of patients with acute ischemic stroke treated with EVT.
Trial registration number
ISRCTN 76741621
AS36-022
ACUTE STROKE TREATMENT BY A PHYSICIAN-BASED EMERGENCY SERVICES TEAM IN A MOBILE STROKE UNIT
1The Norwegian Air Ambulance Foundation, Research, Oslo, Norway
2University of Oslo, Institute of Basic Medical Sciences, Oslo, Norway
3Østfold Hospital Kalnes, Dep. of Neurology, Sarpsborg, Norway
4Oslo University Hospital, Department of Neurology, Oslo, Norway
5University of Stavanger, Faculty of Health Sciences, Stavanger, Norway
Background and Aims
Thrombolytic therapy in acute ischemic stroke is more effective when given early after symptom onset, and different models of Mobile Stroke Units (MSUs) show a reduction in time to treatment in ischemic stroke. In Treat-NASPP (Norwegian Acute Stroke Prehospital Project) we investigate if a non-neurologist physician-based emergency medical services (EMS) team is timesaving and safe in an MSU-system.
Methods
Treat-NASPP is an ongoing, prospective, controlled intervention study. Patients with onset of stroke symptoms within the last 4 hours are included. The MSU-team mimics the Norwegian Helicopter Emergency Medical Services (HEMS) crew, and consists of an anesthesiologist trained in prehospital critical care (air ambulance physician), a nurse and a paramedic. The control group is the ordinary pathway.
Results
Median time (quartiles) from symptom onset to thrombolytic therapy is 98 minutes (72,167) in the MSU (n = 47) compared to 112 (98,140) in the control group (n = 18), p = 0.620.
Median time (quartiles) from dispatch to thrombolytic therapy is 26 minutes shorter in the MSU; 54 (47,66) vs 80 (67,95), p < 0.001.
The golden hour rate is 15% in the MSU vs 0 in the ambulance group, p = 0,175.
Median (quartiles) modified Rankin Scale day 90 is 0 (0-2) in the MSU-group versus 1 (0-4), p = 0,249.
No serious adverse events or symptomatic intracranial hemorrhages have occurred.
Conclusions
Preliminary results indicate that a physician-based EMS crew in a mobile stroke unit, is timesaving and safe.
Trial registration number
NCT03158259 (clinicaltrials.gov)
WITHDRAWN
AS36-012
EFFECTS OF AEROBIC TRAINING ON MEMORY, ATTENTION, AND WORKING MEMORY IN PATIENTS WITH STROKE AND TRAUMATIC BRAIN INJURY
1Div of Rehabilitation Medicine- Danderyd University Hospital, Dept of Clinical Sciences, Stockholm, Sweden
Background and Aims
Patients with stroke or traumatic brain injury (TBI) often suffer from impaired memory, attention deficits and fatigue with related consequences for daily life. Experimental studies show that aerobic training promotes plastic changes in the brain with development of new vasculature, changes in neurotransmitter levels and increase in Brain-Derived Neurotrophic Factor. Furthermore, aerobic training improves cognition in the general population. The effects of training on cognition after stroke and TBI are, however, not well known. We therefore study if aerobic training improves memory, attention and working memory in patients with stroke or TBI.
Methods
This randomized (1:1) controlled study includes 20 patients with cognitive impairments, 18-60 years old. All participate in a rehabilitation program with a multidisciplinary team over the period of ∼8 weeks. The rehabilitation program consists of physical, psychological, cognitive and communicative training according to the patient’s deficits and individual goals. The intervention consists of 30 min aerobic training, 3-4 times a week on a stationary ergometer bicycle at an intensity of 60-80% of estimated maximum heart rate. The control group participates in the regular rehabilitation program.
Participants are tested before and after the program, and at a 3 months follow-up. The cognitive tests include assessment of verbal, visual, working memory and attention. Physical endurance is tested with Åstrand’s sub-maximal endurance test. CNS effects are studied with functional magnetic resonance imaging (fMRI) using blood oxygenation level dependent technique (BOLD).
Results
The study is ongoing. Eighteen patients are so far enrolled. Results will be presented.
Conclusions
N/A
Trial registration number
N/A
AS36-013
THE STROKE-AF RANDOMIZED TRIAL: STROKE OF KNOWN CAUSE AND UNDERLYING ATRIAL FIBRILLATION
1Feinberg School of Medicine of Northwestern University, Davee Department of Neurology, Chicago, USA
2Weill Cornell School of Medicine, Neurology, New York, USA
3Duke Clinical Research Institute, Cardiology, Durham, USA
4Medtronic, Clinical Statistics, Mounds View, USA
5Medtronic, Diagnostics and Monitoring Research, Mounds View, USA
6Massachusetts General Hospital, Neurology, Boston, USA
Background and Aims
Recurrent strokes, particularly those caused by atrial fibrillation (AF), are frequently devastating. Thus, it is important to develop effective secondary stroke prevention strategies. Patients with strokes presumed due to large vessel atherosclerosis or small vessel disease are generally treated with antiplatelet medication. Discovery of AF would usually lead to chronic oral anticoagulation, but its intermittent nature and variable symptom profile can make AF detection difficult. We are comparing two AF detection strategies in this population: continuous arrhythmia monitoring with an insertable cardiac monitor (ICM) vs. standard of care follow-up.
Methods
STROKE-AF is a prospective, randomized, controlled, open-label trial enrolling patients with an ischemic stroke in the past 10 days attributed to large artery atherosclerosis or small vessel disease. Patients must be ≥60 years of age, or 50-59 with ≥1 additional stroke risk factor. Prior AF is exclusionary. Approximately 500 patients at up to 40 centers in the US will be enrolled to assess the primary endpoint which compares AF detection rates through 12 months between patients randomized to ICM or standard of care monitoring. The study is event-driven and powered at > 85% to measure a difference in AF detection rates (≥30 seconds, adjudicated by endpoint committee). Enrollment is ongoing at 29 sites, and 438 subjects have been randomized as of March 22, 2019.
Results

Conclusions
STROKE-AF will provide information on the incidence of AF in patients with an ischemic stroke of presumed known origin and on the utility of long-term continuous monitoring in this population.
Trial registration number
NCT02700945
AS36-014
UPDATE ON THE EFFECTS TRIAL OF FLUOXETINE FOR STROKE RECOVERY: AN INVESTIGATOR-LED RANDOMISED CONTROLLED STUDY IN SWEDEN
1Uppsala university, Department of Neuroscience- Neurology, Uppsala, Sweden
Background and Aims
Animal studies and several small human studies suggest that fluoxetine improves neurological recovery after stroke. In contrast, the most recent and largest fluoxetine study for stroke recovery (FOCUS trial, n = 3,127) was neutral. This abstract presents un update on EFFECTS, a trial of fluoxetine for stroke recovery.
Methods
In this investigator-led, multicentre, parallel group, randomised, placebo-controlled trial, we enrolled non-depressed stroke patients aged 18 years or older between 2-5 days after stroke. Inclusion required a clinical diagnosis of stroke (ischaemic or intracerebral haemorrhage) with persisting focal neurological deficits. Patients were randomly assigned to fluoxetine 20 mg once daily or placebo capsules for 6 months.
The primary outcome is functional status, measured with the modified Rankin scale at the 6-month follow-up, using ordinal logistic regression.
Results
Recruitment began on 20 October 2014. Half of the 1,500 patients were recruited from 20% of the centres (Table). The results will be available within one year.
**TABLE legend: All centres with their Principal Investigator and total numbers of included patients per centre.
Thus is a preliminary table, numbers and correct spelling of the centres needs to be updated ***
Conclusions
The data from the trial will improve the external validity and precision of the estimates of the efficacy and safety of fluoxetine in ischaemic and haemorrhagic stroke.
Trial registration number
EudraCT 2011-006130-16. Registered on 8 August 2014.
ISRCTN, ISRCTN13020412. Registered on 19 December 2014.
ClinicalTrials.gov: NCT02683213, retrospectively registered on 2 February 2016
AS36-015
A RANDOMIZED CONTROLLED TRIAL TO TEST EFFICACY AND SAFETY OF THROMBECTOMY IN STROKE WITH EXTENDED LESION AND EXTENDED TIME WINDOW (TENSION)
1Universitätsklinikum Heidelberg, Neuroradiology, Heidelberg, Germany
2Heidelberg University Hospital, Neuroradiology, Heidelberg, Germany
3Oslo University Hospital, Neurology, Oslo, Norway
4Hospices de Lyon, Biostatistique, Lyon, France
5Karolinska University Hospital, Neuroradiology, Stockholm, Sweden
6Medizinische Universitätsklinik Inssbruck, Neuroradiology, Innsbruck, Austria
7Charles University Hradec Kralove, Neuroradiology, Hradec Kralove, Czech Republic
8Centre Hospitalier Universitaire de Reims, Neuroradiology, Reims, France
9Stroke Alliance for Europe, Stroke Alliance for Europe, London, United Kingdom
10Aarhus University Hospital, Neurologie, Aarhus, Denmark
11Comenius University’s Jessenius Faculty of Medicine and University, Neuroradiology, Martin, Slovak Republic
12Universitätsklinikum Hamburg-Eppendorf, Neuroradiology, Hamburg, Germany
13Universitätsklinikum Hamburg-Eppendorf, Neurology, Hamburg, Germany
Background and Aims
The benefit of thrombectomy in patients with large intracranial vessel occlusion of the anterior circulation has been shown in selected patients in previous randomized controlled trials, but patients with extended stroke lesions were excluded in the majority of these trials. TENSION aims to demonstrate efficacy and safety of thrombectomy in patients with extended stroke lesions in an extended time window or unknown stroke onset.
Methods
TENSION is an investigator-initiated, prospective, open label, blinded endpoint (PROBE), European, two-arm, randomized, controlled, post-market study to compare the safety and effectiveness of thrombectomy as compared to best medical care alone in stroke patients with extended stroke lesions defined by an ASPECTS of 3-5 and in an extended time window (up to 12 hours, or unknown time of symptom onset). In an adaptive design study, up to 714 patients will be randomized.
Primary efficacy endpoint will be clinical outcome defined by the modified Rankin Scale (mRS) at 90 days post-stroke. The main safety endpoint will be death and dependency (mRS 4-6) at 90 days. Additional measures include adverse events, functional health status, post-stroke depression and costs utility assessment.
Results
The trial started randomizing in July 2018. An update on active sites and patient enrollment will be given.
Conclusions
TENSION will provide evidence for effective treatment for severe stroke patients thereby improving functional outcome and quality of life of thousands of stroke patients.
Trial registration number
ClinicalTrials.gov NCT03094715
European Union’s Horizon 2020 grant agreement No 754640
AS36-005
STUDY UPDATE: INTERACT3 – A STEPPED-WEDGE CLUSTER RANDOMIZED TRIAL
1The George Institute for Global Health Australia Beijing Representative Office, stroke, Beijing, China
2West China Hospital- Sichuan University, Neurosurgery, Chengdu, China
Background and Aims
Continued uncertainty exists over benefits of early intensive blood pressure (BP) lowering in acute intracerebral hemorrhage (ICH), related to the non-significant primary outcomes, patient selection, and discordant results of INTERACT2 and ATACH-II. We designed INTERACT3 to determine the effectiveness of a goal-directed care bundle of ‘early intensive physiological control’ (intensive BP lowering, glycemic control, and treatment of pyrexia) and reversal of anticoagulation vs. usual care in ICH.
Methods
INTERACT3 is an international, multicenter, stepped-wedge (4 phases/3 steps), cluster randomized. After a variable background control period, each hospital will implement the intervention (intensive BP lowering – systolic target <140mmHg; glucose control – target 6.1-7.8 mmol/l for non-diabetic, 7.8-10.0 mmol/l for diabetic patients; treatment to a target body temperature ≤37.5 °C; reversal of anticoagulation – target INR <1.5 within 1 hour). After collection of in-hospital clinical/management data and 7-day outcomes, central trained blinded assessors conduct telephone disability assessments (mRS) at 6 months. Primary outcome is an ordinal shift analysis of 180-day mRS scores. Assuming ICC 0.03, the sample size of 75 hospitals, 8661 patients provides 90% power (p = 0.05) to detect a 5.6% absolute improvement (shift) in the primary outcome of the intervention.
Results
No result. It is on-going trial.
Conclusions
INTERACT3 has been approved by West China Hospital Ethics Committee on 27th September 2017 and passed the annual review. To the end of 2018, 1774 ICH patients have been enrolled from 45 Chinese hospitals and 21 sites has shifted into intervention phases with 550 patients undertaking intensive care bundle.
Trial registration number
ChiCTR- IOC-17011787
AS36-006
MORBIDITY PREVALENCE ESTIMATE AT SIX MONTHS FOLLOWING A STROKE: A COHORT STUDY (MORE PRECISE)
1Cardiff University, Division of Population Medicine, Cardiff, United Kingdom
2Anuerin Bevan University Health Board, Research & Development, Newport, United Kingdom
3King’s College London, Department of Biostatistics and Health Informatics- Institute of Psychology Psychiatry and Neuroscience, London, United Kingdom
Background and Aims
Background
The prevalence of morbidity secondary to stroke is of central importance to healthcare professionals, healthcare commissioners, third sector organisations and stroke survivors to understand the likely progress of post-stroke sequelae and to aid in commissioning decisions, care planning and adjusting to life after stroke.
Aims
The aim of the study is to determine the prevalence of morbidity following a stroke, predictors of morbidity and trends using a 15 question Patient Reported Outcome Measure (PROM), cognitive and functional assessments.
Methods
A Wales & England multi-centre cohort study. 500 participants will be recruited across Wales and England within 14 days following an admission to a stroke unit with an ischaemic or haemorrhagic stroke. Participants are assessed at baseline ≤14 days post stroke and subsequently at 90 ( ± 14 days) and 180 ( ± 14 days) post-stroke. At each time point a range of data will be collected relating to the following domains; demographic, routine clinical, patient-reported, cognitive status, emotional well-being and functional ability.
Outcome measures
The primary outcome is the prevalence of morbidity (95%CI) at six months following a stroke. Further analysis will consider temporal changes in the data between the baseline, 3 months and 6 month time points.
Results
N/A
Conclusions
N/A
Trial registration number
Trial Identifier NCT03605381
AS36-001
TR-VENUS: NON-INVASIVE TRANSCUTANEOUS CERVICAL VAGUS NERVE STIMULATION AS A TREATMENT FOR ACUTE STROKE; SAFETY AND FEASIBILITY STUDY
1Hacettepe University Faculty of Medicine, Department of Neurology, Ankara, Turkey
2Massachusetts General Hospital- Harvard Medical School, Radiology, Boston- MA, USA
3electroCore- Inc, electroCore- Inc, Basking Ridge- NJ, USA
Background and Aims
Electrical stimulation of vagus nerve significantly reduces infarct volume and improves functional outcome in experimental stroke models. This translational study will evaluate safety and feasibility of non-invasive transcutaneous cervical vagus nerve stimulation (nVNS) in acute human stroke.
Methods
TR-VENUS is a multi-center, randomized, sham-controlled clinical trial of nVNS in acute stroke. Patients with acute ischemic or hemorrhagic stroke will be randomized into one of the two doses of nVNS or sham stimulation (1:1:1 ratio; n = 20 each) initiated within 6 hours of stroke onset. nVNS will be delivered using a pre-programmed, push-button, hand-held stimulator (gammaCore®) placed on neck. Sham device will produce a buzzing sound without delivering active electrical stimulation. Dose-1 and sham groups will receive 7 consecutive 2-minute trains, one every 10 minutes for 1 hour. Dose-2 group will receive additional 7 consecutive 2-minute trains, one every 10 minutes applied 3 hours after completion of initial dose.
Results
The primary safety endpoint is occurrence of any of the following events: severe bradycardia (HR:≤50/min), or hypotension (≥20 mmHg reduction in MAP) during stimulation, progression of neurologic deficit (NIHSS score: ≥4 points increase), or death within 24 hours of treatment. Additional data will be collected on feasibility (proportion of eligible patients receiving nVNS within 6 hours, proportion completing all pre-specified doses, and stroke onset-to-nVNS time) and efficacy (infarct growth at 24 hours) end-points. Statistical analysis will be performed by means of descriptive analytical methods.
Conclusions
This study will provide key data to help evaluate the therapeutic potential of nVNS in acute stroke in humans.
Trial registration number
ClinicalTrials.govIdentifier:NCT03733431
AS36-018
ESMOLOL FOR THE TREATMENT OF HYPERTENSION AFTER INTRACEREBRAL HEMORRHAGE STUDY – ETHICHS
1Ribeirão Preto Medical School – University of São Paulo, Neurosciences and Behavioral Sciences, Ribeirão Preto, Brazil
2Ribeirão Preto Medical School – University of São Paulo, Internal medicine, Ribeirão Preto, Brazil
3Federal University of São Paulo, Neurology, São Paulo, Brazil
4Hospital de Clinicas de Porto Alegre – Federal University of Rio Grande do Sul, Medicina Interna, Porto Alegre, Brazil
5Hospital das Clínicas da Faculdade de Medicina de Botucatu – UNESP, Neurology, Botucau, Brazil
6Hospital Geral de Fortaleza, Neurology, Fortaleza, Brazil
7Hospital Madre Teresa, Neurology, Belo Horizonte, Brazil
8Hospital São Rafael, Neurology, Salvador, Brazil
9Cristalia Produtos Químicos Farmacêuticos, Clinical Research Division, São Paulo, Brazil
Background and Aims
Strategies that improve variability of blood pressure during the acute intracerebral haemorrhage (ICH) have potential for controlling hematoma expansion and improve patient prognosis. Labetalol and nicardipine are not available in many developing countries, where sodium nitroprusside has been the main alternative, despite its potential for cyanide toxicity, antiplatelet effects and risk of increased intracranial pressure. Because of its short half-life and safety profile, esmolol hydrochloride (beta-blocker) is suitable for continuous venous infusion but has never been formally tested in ICH patients. We aim to evaluate the beneficial effects of combining esmolol hydrochloride with sodium nitroprusside for the blood pressure control of participants with ICH.
Methods
prospective, phase IV, multicentre, randomized, open-label, blinded endpoint assessment (PROBE) study. Enrolling 80 patients with an acute supratentorial ICH (diagnosis confirmed by computed tomography or magnetic resonance imaging), and: systolic pressure (SBP) > 150 mmHg, not contraindicated for treatment with beta-blockers, treatment within 6 hours of the stroke, target of ≤ 140 mmHg of SBP within 1-hour after treatment. Primary outcome: variation of systolic and diastolic pressure during the first 7 days measured by intermittent blood pressure monitoring. Secondary outcomes include the percentage of participants with controlled SBP, distribution of the mRs scores at 90-days and non-invasive assessment of intracranial pressure using optic nerve sheet diameter measured by ultrasound.
Results
Study is currently enrolling patients in 8 centres in Brazil.
Conclusions
we aim to investigate the potential benefits of using esmolol hydrochloride infusion for blood pressure control in patients with acute ICH.
Trial registration number
NCT number: NCT03743103
AS36-019
A PHASE III, PROSPECTIVE, DOUBLE-BLIND, RANDOMIZED, PLACEBO-CONTROLLED TRIAL TO ASSESS THE EFFICACY AND SAFETY OF TENECTEPLASE IN IMAGING-ELIGIBLE, LATE-WINDOW PATIENTS WITH ACUTE ISCHEMIC STROKE (TIMELESS)
1Stanford University Medical Center, Neurology and Neurological Sciences, Stanford, USA
2UC Gardner Neuroscience Institute, Neurology, Cincinatti, USA
3Univeristy of Alberta, Neurology, Edmonton, Canada
4Melbourne Braine Centre at the Royal Melbourne Hospital, Medicine and Neurology, Parkville, Australia
5Vanderbilt Cerebrovascular Program, Neurology, Nashville, USA
6Stanford University, Neurosciences Institute, Palo Alto, USA
7Neurovascular Imaging Research Core and UCLA Stroke Center, Neurology, Los Angeles, USA
8Hartford Hospital, Neurology, Hartford, USA
9Massachusetts General Hospital, Neurology, Boston, USA
10Genentech Inc., US Medical Affairs, South San Francisco, USA
Background and Aims
Systemic thrombolysis with alteplase, a human tissue plasminogen activator (tPA), is standard of care for eligible patients ≤4.5 hours of acute ischemic stroke (AIS). For patients with AIS due to large vessel occlusion (LVO) with a salvageable brain pattern on advanced imaging, mechanical thrombectomy (MT) ≤24 hours after AIS onset improves functional outcomes. Tenecteplase, a modified tPA, has shown potential for treating > 4.5 hours and may achieve reperfusion before MT.
Methods
This Phase III, prospective, double-blind, randomized, placebo-controlled trial will evaluate the efficacy and safety of tenecteplase vs placebo in patients with AIS (age ≥18 years) presenting in the 4.5–24-hour time window, who have LVO and meet CT or MR perfusion mismatch imaging criteria with ischemic core <70 mL (NCT03785678;
Results

Conclusions
This trial will determine if treatment with IV tenecteplase in patients with AIS who have LVO and salvageable brain tissue on imaging 4.5–24 hours after onset results in improved clinical outcomes, including early reperfusion that may reduce the need for endovascular therapy.
Trial registration number
NCT03785678
AS36-020
HOW DOES THE DESIGN OF INPATIENT REHABILITATION FACILITIES INTERACT WITH STROKE CARE?: STUDY DESIGN AND PROTOCOL
1The Florey Institute of Neuroscience and Mental Health, Stroke, Heidelberg, Australia
2NHMRC Centre for Research Excellence in Stroke Rehabilitation and Brain Recovery, Heidelberg, Victoria, Australia
3Griffith University, Menzies Health Institute Queensland, Brisbane, Australia
4The Hopkins Centre Research for Rehabilitation and Resilience, Brisbane, Queensland, Australia
Background and Aims
Hospital building design can impact staff and patient safety, clinical outcomes, economic performance, and emotional well-being of all users. The majority of healthcare design evidence comes from research in acute healthcare settings. Stroke rehabilitation differs from acute care in many ways, but there are no building design recommendations specific to this population. The aim of this study is to explore how the built environment may best support efficiency, clinical outcomes, emotional well-being, and safety in inpatient stroke rehabilitation.
Methods
This research will follow a Convergent Mixed-Methods study design within a multiple-case study framework. Data will be collected in two inpatient rehabilitation facilities in Victoria, Australia, between November 2018 and April 2019. Figure 1 details the qualitative and quantitative data that will be collected and how this will address the aim of the study. Twenty staff at each site will be interviewed, and 20 patients at each site will participate in the behavioural mapping, questionnaires, and interview. The “walk-through” semi-structured interviews will be analysed using a Qualitative Descriptive approach. The qualitative and quantitative data will be collected in parallel, analysed separately, and then merged to produce a multiple-case study report.
Results
This poster presents further detail about the study protocol and progress.
Conclusions
This protocol presents a new method for researching the interaction between the built environment and stroke care. The results of the study will be used to produce recommendations for the design of inpatient stroke rehabilitation facilities.
Trial registration number
N/A
AS36-008
NOVEL CLINICAL TRIAL DESIGN: INTERACT4, AN AMBULANCE-DELIVERED PROBE TRIAL OF INTENSIVE BP LOWERING
1The George Institute China- Peking University Health Science Center- Beijing- China- The George Institute for Global Health- UNSW- Sydney- Australia, Stroke Division, Shanghai, China
2The First Affiliated Hospital of Chengdu Medical College- China, Department of Neurology, Chengdu, China
3Shanghai Eastern Hospital- Tongji University- China, Department of Neurology, Shanghai, China
4The George Institute for Global Health- UNSW- Sydney- Australia, Academic Project Operation, Sydney, Australia
5The George Institute China- Peking University Health Science Center- Beijing- China, Stroke Division, Shanghai, China
6The George Institute for Global Health- UNSW- Sydney- Australia- Department of Preventive Medicine and Public Health- Faculty of Medicine- Fukuoka University- Fukuoka- Japan, Neurological and Mental Health Division, Sydney, Australia
7The George Institute China- Peking University Health Science Center- Beijing- China- The George Institute for Global Health- UNSW- Sydney- Australia- Department of Neurology- Royal Prince Alfred Hospital- Sydney- Australia- Sydney Medical School- University of Sydney- Sydney- Australia, Executive Office, Beijing, China
Background and Aims
Uncertainty persists over benefits of early intensive blood pressure (BP) lowering both in acute intracerebral hemorrhage (ICH) and acute ischemic stroke (AIS), with speed and choice of treatment being potentially critical factors. The INTERACT4 (ClinicalTrials.gov: NCT03790800) aims to determine if hyperacute ambulance-delivered intensive BP control improves functional outcome after stroke.
Methods
A region-based clustered multicenter, prospective, open, randomized, blinded endpoint trial of pre-hospital BP lowering in 3116 hypertensive presumed acute stroke patients in China. Up to two bolus doses of intravenous Urapidil will be used to treat patients assigned to intensive group (systolic BP target <140mmHg within 30 mins) via a mobile-APP-based randomization system.
Results
Control BP management is according to guidelines after admission to hospital. Data are collected in-hospital over 7-days, and at 90-days through central blind telephone follow-up assessment. Sample size allows 90% power to detect 22% relative odds improvement in functional recovery among 2072 cases of definite stroke (30% mimics) and 30% absolute reduction in 24 hr hematoma growth in ICH patients (30% of definite strokes).
Conclusions
INTERACT4 will provide reliable evidence on the effectiveness and safety of ambulance-delivered intensive BP lowering with a standard intravenous regime in patients with presumed acute stroke.
Trial registration number
ClinicalTrials.gov: NCT03790800
AS36-023
PURINES FOR RAPID IDENTIFICATION OF STROKE MIMICS (PRISM): A DIAGNOSTIC ACCURACY STUDY
1Newcastle University, Stroke Research Group, Newcastle upon Tyne, United Kingdom
2Warwick University, School of Life Sciences, Warwick, United Kingdom
3Oxford University, Medical Sciences Division, Oxford, United Kingdom
4Newcastle upon Tyne Hospitals NHS Foundation Trust, NIHR Newcastle Medtech and In Vitro Diagnostics Co-operative, Newcastle upon Tyne, United Kingdom
5University Hospitals Coventry and Warwickshire NHS Trust, Vascular Surgery, Coventry, United Kingdom
6Newcastle University, NIHR Newcastle Medtech and In Vitro Diagnostics Co-operative, Newcastle upon Tyne, United Kingdom
7Keele University, Institute for Science and Technology in Medicine, Keele, United Kingdom
8Manchester University, Division of Cardiovascular Sciences, Manchester, United Kingdom
Background and Aims
Ambulance personnel rely upon quick but basic symptom checklists to identify stroke such as the Face Arm Speech Test (FAST). These have reasonable sensitivity (70%) but poor specificity (40-70%). Consequently at least 30% of suspected stroke cases later receive a ‘stroke mimic’ diagnosis and require care in settings other than a hyperacute stroke unit (HASU).
Purines are short half-life by-products from energy producing metabolic pathways which accumulate rapidly during hypoxic tissue injury. In the clinical context of suspected stroke, a significant purine difference is expected for confirmed stroke relative to those common mimic conditions where hypoxic tissue is not involved in the underlying mechanism of illness.
This study is evaluating whether a portable point of care purine assay using a finger-prick blood sample (SMARTChip; Sarissa Biomedical Ltd, UK) can differentiate between stroke and mimic conditions during emergency ambulance assessment.
Methods
Study design
Blinded prospective observational cohort study.
Setting
Three regional ambulance services and selected receiving HASUs.
Study population
Adults believed to be with 4 hours of suspected stroke onset when assessed by ambulance personnel.
Index test: SMARTChip Purine assay.
Reference standard tests: Expert clinician opinion informed by brain imaging and/or other investigations will assign the following diagnoses at day 7 after admission: ischaemic stroke, haemorrhagic stroke, TIA and stroke mimic conditions.
Analyses: Sensitivity, specificity, negative and positive predictive values, area under the Receiver Operating Characteristic curve for identification of stroke mimic conditions.
Sample size: 958 participants.
Results
Study progress: Currently in set up with recruitment anticipated to start in April 2019.
Conclusions
n/a
Trial registration number
Not yet registered
AS36-025
SENSITIVE BEDSIDE ASSESSMENT OF SPATIAL NEGLECT, CLINICAL CHARACTERISTICS AND OUTCOMES OF ADULT STROKE PATIENTS: A CROSS-COUNTRY STUDY BETWEEN KAUNAS (LITHUANIA) AND REYKJAVIK (ICELAND)
1University of Iceland, Faculty of Nursing, Reykjavik, Iceland
2Landspitali- The National University Hospital of Iceland, Neurology, Reykjavik, Iceland
3Lithuanian University of Health Sciences, Faculty of Medicine, Kaunas, Lithuania
Background and Aims
Spatial neglect (SN) is common in stroke patients with right hemisphere strokes but remains poorly identified in clinical care. Sensitive methods to assess and detect SN in stroke wards is needed.
Methods
1) Test a novel assessment tool for SN, 2) evaluate the diagnostic accuracy of star cancellation and figure copying between patients with left, right and bilateral hemispheric strokes, by using the Catherine Bergego Scale as a reference standard, 3) determine reasons for inability to complete SN assessment, and 4) compare clinical characteristics, stroke severity, and outcomes between patients with and without SN using; Barthel Index, community mobility, description of helping aids, length of hospital stay, mRS, NIHSS at admission, and patients reported outcome measures (PROMIS-10). Data will be collected at three timepoints, t1: within 5 days following stroke, t2: 3 months after stroke, and t3: at 1-year follow-up. We plan to include 200 patients with SN at baseline out of consecutively admitted stroke patients in Kaunas and Reykjavík.
Results
Participants will be enrolled from June 2019 and until June 2021. ROC-curves will provide assessment of individual test´s ability to differentiate between SN and non-SN. Principal component analysis and Rash analysis will be used to examine inner structure, validity, reliability, linearity and unidimentionality of the novel assessment tool. Descriptive and predictive statistics will be employed to compare clinical characteristic between subgroups of stroke patients and different timepoints.
Conclusions
Pragmatic and effective ways of assessing SN bedside within the stroke ward are expected from the study results.
Trial registration number
N/A-should be available before the conference
AS36-026
TREATMENTSTRATGY IN ACUTE LARGE VESSEL OCCLUSION: TRANSPORT STRATEGY IN PATIENT WITH LARGE VESSEL OCCLUSION? PRIORITIZE IV OR ENDOVASCULAR TREATMENT. A RANDOMIZED CONTROLLED TRIAL
1Aarhus University Hospital, Neurology, Aarhus, Denmark
2Danish Center for Clinical Health Services Research-Aalborg University and Aalborg University Hospital, Department of Clinical Medicine, Aalborg, Denmark
3Pre-hospital Central Denmark Region, Research department, Aarhus, Denmark
4Amsterdam UMC- location AMC, Department of Radiology and Nuclear Medicine, Amsterdam, The Netherlands
5Beth Israel Deaconess Medical Center- Harvard Medical School, Department of Neurology, Boston, USA
Background and Aims
Introduction
Endovascular treatment (EVT) is superior to intraveneous thrombolysis (IVT) in acute ischemic stroke (AIS) patients with large vessel occlusion (LVO).
If a severe stroke with symptoms indicating LVO occurs in the catchment area of a primary stroke center (PSC) evidence is lacking regarding the optimal transport strategy for patients in the IVT-window. Should we bypass PSC in order to get fast EVT at a comprehensive stroke center (CSC)?
Aim
We hypothesize that bypassing the PSC will result in improved 90-day functional outcome for patients with suspected LVO.
Results
Outcomes: The primary outcome will be the 90-day modified Rankin Scale score (mRS) for all patients with AIS. Secondary outcomes include 90-day mRS for all randomized patients, all patients with ischemic stroke but without LVO and for patients with hemorrhagic stroke. The safety outcomes include severe dependency or death and time to IVT for AIS patients.
Conclusions
We assume that study results will influence decision making regarding transport strategy for patients with suspected LVO.
Trial registration number
ClinicalTrials.gov Identifier: NCT03542188
AS36-027
PREHOSPITAL TRIAGE IN ACUTE STROKE: THE PRESTO STUDY
1Albert Schweitzer Hospital, Neurology, Dordrecht, The Netherlands
2Erasmus MC University Medical Center, Neurology, Rotterdam, The Netherlands
3Erasmus MC University Medical Center, Public Health, Rotterdam, The Netherlands
4Maasstad Hospital, Neurology, Rotterdam, The Netherlands
5Franciscus Gasthuis & Vlietland, Neurology, Rotterdam, The Netherlands
6Ambulance Service Rotterdam Rijnmond, Ambulance Service, Barendrecht, The Netherlands
7Ambulance Service Zuid-Holland Zuid, Ambulance Service, Papendrecht, The Netherlands
8Erasmus MC University Medical Center, Radiology and Nuclear Medicine, Rotterdam, The Netherlands
Background and Aims
Only a subset of acute ischemic strokes is caused by an intracranial large vessel occlusion (LVO) in the anterior circulation. The effect of mechanical thrombectomy in these patients declines strongly over time. Prehospital stroke scales are proposed to identify patients with a high likelihood of having an LVO to transport them directly to endovascular-capable centers. We aim to validate several prehospital stroke scales prospectively to assess their accuracy in predicting LVO in the prehospital setting.
Methods
PRESTO is a multi-center observational prospective cohort study in the Netherlands including suspected stroke patients transferred with the ambulance. Paramedics will assess a combination of items from five prehospital stroke scales (LAMS, RACE, C-STAT, PASS, G-FAST). At least 100 patients with an LVO are required for validation of the different stroke scores. Primary outcome is the clinical diagnosis of an acute ischemic stroke with an intracranial LVO in the anterior circulation. Additional hospital data relating to treatment and diagnosis will be collected. Predictive performance of the prehospital stroke scales will be assessed using logistic regression analysis and will be expressed as sensitivity, specificity and area under the receiver operator curve (AUC).
Results
Inclusions started August 2018. At this moment, 428 patients are registered.
Conclusions
End of enrollment is estimated August 2019. The best performing scale, or the simplest scale in case of equivalence, will be integrated in a decision model with clinical characteristics and ambulance driving times to improve prehospital triage of suspected stroke patients.
Trial registration number
NTR7595 (www.trialregister.nl)
AS36-004
STUDY DESIGN AND RESULTS OF THE PILOT PHASE OF A CLUSTER-RANDOMIZED MULTIMODAL POST-STROKE CARE INTERVENTION TRIAL – THE AMBULATORY STRUCTURED POST-STROKE CARE PROGRAM (SANO)
1University of Würzburg, Institute for Clinical Epidemiology and Biometry, Würzburg, Germany
2Klinikum der Stadt Ludwigshafen a. Rh., Neurological Clinic, Ludwigshafen, Germany
3Bezirkskrankenhaus Günzburg, Clinic for Neurology and Neurological Rehabilitation, Günzburg, Germany
4Benedictus Krankenhaus Tutzing, Neurology, Tutzing, Germany
5Charité Universitätsmedizin Berlin, Department of Neurology with Experimental Neurology, Berlin, Germany
6Klinikum der Universität München, Institute for Stroke and Dementia Research, München, Germany
7Universitätsklinikum Würzburg, Neurological Clinic and Policlinic, Würzburg, Germany
8Universitätsmedizin der Johannes-Gutenberg-Universität, Clinic and Policlinic for Neurology, Mainz, Germany
9German Stroke Society, Stroke Unit Zertifizierungsausschuss, Berlin, Germany
10Bayerischer Hausärzteverband, General Practitioner, Würzburg, Germany
11Universitätsklinikum Jena, Institute for Medical Statistics- Computer Science and Data Science, Jena, Germany
12Westfälische Hochschule Gelsenkirchen, Institute for Work and Technology, Gelsenkirchen, Germany
Background and Aims
Previous studies from routine clinical care in Germany showed deficits in cardiovascular risk factor management among stroke patients and high rates of recurrent events and rehospitalization after discharge.
The SANO trial will investigate whether an evidence-based cross-sectoral post-stroke care program significantly reduces the rate of the combined endpoint recurrent stroke, myocardial infarction and death within the first year after ischemic stroke.
Methods
SANO is a parallel-arm cluster-randomized trial intended to recruit 2,790 patients across 30 regions in Germany. Development of the intervention (Figure) followed the recommendations of the Medical Research Council Framework for complex interventions, combining both structural and patient-centered elements. The study was piloted in two regions prior to study start.

Results
All regions were randomized and recruitment started in January 2019. Eighteen patients were included in the pilot study (mean age 66 ± 12 years; 72% men). Recruitment rate among all hospitalized stroke patients was 12%. Average duration of the baseline assessment was 128 ± 46 min and the vast majority of patients reported to be satisfied or very satisfied with its organization (98.8%) and duration (91.7%).
Conclusions
Baseline assessment of the intended post-stroke care program was feasible. Enrolment is expected to be finished in January 2020 and primary results of the SANO trial will be reported approximately in July 2021.
Trial registration number
DRKS00015322
AS36-028
TRANSESOPHAGEAL ECHOCARDIOGRAPHY – DYSPHAGIA RISK IN ACUTE STROKE -T.E.D.R.A.S.- TRIAL
1University Hospital Giessen and Marburg, Department of Neurology, Giessen, Germany
Background and Aims
Dysphagia is common among acute stroke patients. Transesophageal echocardiography (TEE) is routine in examining causes of cardiac strokes, and the incidence of dysphagia among acute stroke patients undergoing TEE is unknown.
Methods
Using flexible endoscopic evaluation of swallowing (FEES), T.E.D.R.A.S., as a prospective, double-blinded, randomized, and controlled study, assesses whether TEE increases the risk of dysphagia among acute stroke patients. Preliminary results of 29 patients are presented. All patients (intervention and control group) underwent FEES at three different time points (24 hours before, immediately after, and 24 hours after TEE). The Penetration-Aspiration-Scale (PAS), Secretion Severity Rating Scale (SSRS), and the Yale Pharyngeal Residue Severity Rating Scale (YPRSRS) assessed the severity of airway invasion during swallowing and hypopharyngeal residue. Difference scores were built from pre to post TEE for all dysphagia measures following between group comparisons with Mann-Whitney U-test.
Results
Compared to the control group, the intervention group shows an increase in dysphagia measures immediately after TEE (p = 0.009 for PAS-saliva, p = 0.047 for SSRS) and 24 hours after TEE (p = 0.032 for SSRS). Compared to the control group, the intervention group shows an increase of residue severity in the YPRSRS for valleculae immediately after TEE (p = 0.028 for small liquid bolus; p = 0.04 for large liquid bolus) and 24 hours after TEE (p = 0.001 for small liquid bolus; p = 0.02 for large liquid boluses).
Conclusions
The preliminary data of the T.E.D.R.A.S. trial indicate that TEE may have a negative influence on swallowing ability among acute stroke patients.
Trial registration number
223/12
AS36-029
STROKE AND FABRY DISEASE: PROTOCOL OF AN ITALIAN MULTICENTER, PROSPECTIVE, OBSERVATIONAL, LONGITUDINAL STUDY (THE “RIFS ” REGISTRY)
1University of Florence, NEUROFARBA, Florence, Italy
2AOU Careggi, Dea, Florence, Italy
Background and Aims
Fabry disease (FD) is an often unrecognized cause of both ischemic and hemorrhagic strokes. Prevalence hitherto reported in stroke populations is not negligible. Data are scanty about characteristics of patients with stroke due to FD. In the framework of a nationwide Italian dedicated registry, we planned to prospectively screening for FD young (<60 y.o.) stroke/TIA patients consecutively admitted after a cerebrovascular event. Primary objective is the assessment of stroke-recurrence rate and neurological or multi-systemic outcomes among FD-related stroke patients. Secondary objectives are i) identification of hallmarks which could help to improve FD diagnosis among stroke patients, ii) investigation of potential co-factors of stroke in patients with FD, and iii) comparison of outcomes among FD patients treated with ERT vs untreated patients.
Methods
Data are being web-based registered about risk factors, FD markers, stroke pathogenesis (classified through CCS or SMASH-U), neuroimaging, alfa-galactosidase and GLA-gene results. In patients eventually diagnosed with FD (and their FD relatives), further records about multi-system involvement and neurological outcomes are being collected at baseline, 6 and 12 months. This project was funded by Shire International GmbH under IIR-ITA-000977.
Results
We aim to screen 1700 eligible stroke patients in 38 acute neurological stroke units across Italy. We are waiting to identify up to 48 new FD-related strokes (and 240 FD relatives). Recruitment will be closed in March 2020.
Conclusions
Products of the RIFS Registry will include, besides systematic recording of outcomes in FD-related strokes, an analysis on a large number of aspects of stroke etiology in young-adult patients.
Trial registration number
CEAVC10311
AS36-030
INFLUENCE OF A PHYSICAL EXERCISE PROGRAM ON CARDIAC REMODELING AND ON FUNCTIONAL CAPACITY OF PATIENTS WITH STROKE
1Sao Paulo State University UNESP, Department of Internal Medicine, Botucatu, Brazil
2Federal University of Triângulo Mineiro UFTM, Department of Applied Physical Therapy, Uberaba, Brazil
3Sao Paulo State University UNESP, Department of Neurology- Psychology and Psychiatry, Botucatu, Brazil
Background and Aims
Cardiovascular rehabilitation can improve the functional independence after stroke. The aim was to verify the effect of a physical exercise program on the echocardiographic variables in patients with chronic ischemic stroke (IS) and to evaluate the functional capacity (FC) and quality of life (QL) after the intervention.
Methods
Clinical, randomized, blind trial, with 40 patients from 6 to 12 months after IS, clinically stable, both sexes and <18 years old. Control Group (n = 20): conventional physiotherapeutic intervention following the standard protocol according to the National Institute for Health and Care Excellence. Intervention Group (n = 20): cardiovascular rehabilitation on a treadmill programmed at speed and inclination compatible with individual capacity, 45-minute duration, three times per week, for 16 weeks. The groups will be evaluated initially and at the end, by the 6-minute walking test; FC by the NIHSS score, mRs and Barthel Index; nutritional evaluation by electric bioimpedance; ambulatory blood pressure monitoring, transthoracic echocardiogram and QL by EUROQOL.
Results
Since August/2017, 189 patients were diagnosed with IS, 151 were excluded and 28 recruited, of which 22 were randomized and protocol initiated while 6 await randomization. The greatest difficulties until the present include difficulty of transportation to the study center, patient’s unavailability from the intervention group for attending three times per week and disinterest in participating in the study due to cultural and socioeconomic aspects of this population.
Conclusions
The cardiovascular rehabilitation program is expected to have a favorable impact on the aerobic capacity and quality of life of patients after stroke.
Trial registration number
RBR-4wk4b3
AS36-031
LONGITUDINAL STUDY OF YOUNG PATIENTS WITH EMBOLIC STROKE OF UNDETERMINED SOURCE (ESUS)
1Mcmaster University, Medicine Neurology, Hamilton, Canada
Background and Aims
Stroke in young adults is not rare, and can have a devastating, lasting impact. Up to 20% of patients with Embolic Stroke of Undetermined Source (ESUS) are under 50 years of age; thus, determining potential causes and outcomes in this younger cohort may significantly impact clinical practice. We aim to describe clinical, laboratory and imaging characteristics of patients between 21 and 50 years of age with ESUS; determine rates of new-onset atrial fibrillation; and investigate predictors of recurrent stroke in this unique population.
Methods
This is an ongoing multi-center, international registry, which plans to prospectively enroll 500 patients between the ages of 21-50 years with ESUS within 60 days. Clinical, laboratory and imaging data are documented at enrollment. Patients will be followed prospectively at 6, 12 and 18 months post-stroke via telephone interview to determine treatment and outcomes. End of enrollment is anticipated in July 2019.
Results
There were 45.6% females enrolled in the cohort. Median age at stroke was 43 years. IV-tPA was given to 54/296 (18.4%). Median modified Rankin Score at time of stroke was 1.0 (range 0-5.0). 132/290 (45.5%) of patients had a PFO by either TTE or TEE. Hypertension (24.7%) and current tobacco smoking (24.7%) was the most common risk factor.
Table 1 contains baseline characteristics and diagnostic workup of 304/338 patients enrolled thus far.
Conclusions
Demographics, stroke risk factors and recurrence rates may differ for younger patients with ESUS; thus, findings of this study could potentially shape clinical practice and ultimately improve outcomes for this at-risk population.
Trial registration number
N/A
AS36-032
DUAL THROMBOLYTIC THERAPY WITH MUTANT PRO-UROKINASE AND LOW DOSE ALTEPLASE FOR ISCHEMIC STROKE (DUMAS)
1Erasmus MC University Medical Center, Neurology, Rotterdam, The Netherlands
2Erasmus MC University Medical Center, Radiology and Nuclear Medicine, Rotterdam, The Netherlands
3Reinier de Graaf Group, Neurology, Delft, The Netherlands
4Haaglanden Medical Center, Neurology, The Hague, The Netherlands
5Albert Schweitzer Hospital, Neurology, Dordrecht, The Netherlands
Background and Aims
The effectiveness of alteplase is limited and the occurrence of intracranial and extracranial hemorrhage is a major limitation. Dual thrombolytic therapy with a low dose alteplase followed by mutant pro-urokinase (m-pro-urokinase), which does not lyse hemostatic fibrin, has a significant potential to be safer and more efficacious. Our aim is to test the safety and preliminary efficacy of dual thrombolytic treatment consisting of a bolus alteplase followed by m-pro-urokinase against usual treatment with alteplase in patients presenting with ischemic stroke.
Methods
DUMAS is a multicenter, phase II, randomized clinical trial with open-label treatment, adaptive design for dose optimization and blinded outcome assessment. We will enroll 200 patients with a clinical diagnosis of ischemic stroke and who meet the criteria for standard treatment with intravenous (IV) alteplase. Patients eligible for endovascular thrombectomy will be excluded. Patients will be randomly assigned (1:1) to receive a bolus of IV alteplase (5mg) followed by continuous IV infusion of m-pro-urokinase (40mg/hr during 60 minutes) or usual care with alteplase (0.9mg/kg). The primary outcome will be any post-intervention intracranial hemorrhage on MRI according to the Heidelberg Bleeding Classification within 24-48 hours. Secondary outcomes include symptomatic intracranial hemorrhage, change (pre-treatment vs. post-treatment) in abnormal perfusion volume and score on the mRS at 90 days.
Results
IRB approval is pending.
Conclusions
We hypothesize that dual thrombolytic therapy with a bolus alteplase followed by m-pro-urokinase will reduce the occurrence of intracranial hemorrhage in patients with ischemic stroke compared to patients treated with alteplase alone.
Trial registration number
NTR7634 (www.trialregister.nl)
AS36-033
NONINVASIVE CEREBRAL ELECTRICAL STIMULATION TO REDUCE UNILATERAL SPATIAL NEGLECT AFTER STROKE: PARTIAL RESULTS OF ELETRON TRIAL
1Sao Paulo State University UNESP, Rehabilitation Department, Botucatu, Brazil
2Sao Paulo State University UNESP, Department of Internal Medicine, Botucatu, Brazil
3Sao Paulo State University UNESP, Department of Neurology- Psychology and Psychiatry, Botucatu, Brazil
4Sao Paulo University USP, Department of Neurology, São Paulo, Brazil
5University of Toronto, Department of Surgery, Toronto, Canada
6Federal University of Triângulo Mineiro UFTM, Department of Applied Physical Therapy, Uberaba, Brazil
Background and Aims
Balance disturbances of brain electrical activity can cause Unilateral Spatial Neglect (USN) after stroke. Transcranial direct-current stimulation (tDCS) can improve the USN prognosis by rebalancing hemispheric activity. The aim was to describe the USN evolution in individuals submitted to tDCS after stroke.
Methods
Randomized, controlled, double-blind clinical trial with≥18 years old patients, both genders, with stroke and USN diagnosed by Behavioural Inattention Test (BIT). Randomization to three groups: 1 – Anodic tDCS in right parietal lobe; 2 – Cathodic tDCS in left parietal lobe; 3 – Control (sham). All patients receive conventional physiotherapy after tDCS (15 sessions). USN was assessed by Catherine Bergego Scale (CB) and BIT by a blind investigator on day 1 (A1), and on the last treatment day (A15). Groups were compared using non-parametric Kruskal-Wallis and Fisher’s exact tests. Significance level 5%.
Results
On a preliminary analysis, 22 patients were evaluated up to the present moment. The BIT and CB scores showed USN improvement in the 3 groups, a tendency for significance was found in CB score (Table 1).

Conclusions
Partial results suggests that physical rehabilitation has an influence on USN, it isn’t yet possible to assess whether the tDCS potentiates this result. It seems that the anodic stimulation treatment is superior for the NEU treatment.
Trial registration number
REBEC – RBR-78jvzx
AS36-007
CAROTID REVASCULARIZATION AND MEDICAL MANAGEMENT FOR ASYMPTOMATIC CAROTID STENOSIS: CREST-2 UPDATE
1Mayo Clinic, Department of Neurology, Jacksonville, USA
2University of Maryland, Vascular Surgery, Baltimore, USA
3The Univesity of Alabama at Birmingham, Department of Biostatistics, Birmingham, USA
4Medical University of South Carolina, Department of Neurology, Charleston, USA
5Cardiovascular Associates of the Southeast, Interventional Cardiology, Birmingham, USA
6Mayo Clinic, Department of Neurology, Rochester, USA
7University of Maryland, Department of Neurology, Baltimore, USA
8Mayo Clinic, Department of Neurology, Scottsdale, USA
9Mayo Clinic, Department of Radiology, Rochester, USA
10University of California at Los Angeles, Vascular Surgery, Los Angeles, USA
11National Institute of Neurological Disorders and Stroke, Division of Clinical Research, Rockville, USA
Background and Aims
The applicability of prior randomized trials in the management of asymptomatic carotid stenosis to current treatment decisions has been questioned. The NINDS-funded CREST-2 will compare CEA and intensive medical management (IMM) versus IMM alone (n = 1240), and CAS and IMM versus IMM alone (n = 1240) in asymptomatic patients with ≥70% stenosis.
Methods
CREST-2 consists of two parallel randomized clinical trials at ≈120 centers, including collaboration with NINDS StrokeNet. The composite primary outcome is stroke or death during the peri-procedural period or ipsilateral ischemic stroke thereafter up to 4 years. Cognitive status will be assessed periodically through computer-assisted telephone interviews. Centrally directed IMM includes tight control of blood pressure (systolic target <130 mm Hg) and cholesterol (LDL target <70 mg/dl) as well as lifestyle coaching.
Results
As of January 22, 2019, 185 centers have been approved by the Site Selection Committee. Credentialing is ongoing, with 403 approved surgeons and 182 approved interventionists. An additional 118 interventionists have been approved to submit additional cases via the CREST-2 Companion Registry which provides a CMS-reimbursed pathway for full credentialing in CREST-2. 1285 patients have been randomized, 667 (52%) patients in the endarterectomy trial, and 618 (48%) patients in the stent trial.
Conclusions
CREST-2 is designed to identify the best approach for asymptomatic carotid stenosis. An update will be provided regarding the numbers of patients randomized, centers certified, as well as surgeons and interventionists fully approved. The CREST-2 Registry will provide the option of CAS while enhancing interventionists’ credentials for participation in CREST-2.
Trial registration number
NS080168
AS36-009
TREAT_CCM CLINICAL TRIAL – A MULTICENTER RANDOMIZED CLINICAL TRIAL
1IRCCS Foundation C. Besta Neurological Institute, Cerebrovascular Unit, Milan, Italy
2Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico- Milan- Italy, Department of Neurology, Milan, Italy
3Centro Neurolesi Bonino Pulejo- Messina- Italy, Centro Neurolesi Bonino Pulejo, Messina, Italy
4Università Cattolica del Sacro Cuore, Department of Neurosurgery, Rome-, Italy
5Ospedale Niguarda, Centro Munari Chirurgia dell’Epilessia e del Parkinson, Milan, Italy
6Fondazione IRCCS Casa Sollievo della Sofferenza, Medical Genetic Unit, San Giovanni Rotondo, Italy
7Istituto di Ricerche Farmacologiche “Mario Negri”, Department of Cardiovascular Research, Milan, Italy
8IFOM- Firc Institute for Molecular Oncology, Laboratory of Vascular Biology, Milan, Italy
Background and Aims
Cerebral cavernous malformations (CCM) is a cerebrovascular disease characterized by the presence of isolated or multiple CCM lesions, causing recurrent headache, seizures, focal neurological deficits and hemorrhages. Anecdotal reports support the efficacy of the non-selective beta-blocker propranolol in CCM. The primary objective of this trial is to test whether a chronic treatment with propranolol will reduce the burden of cerebrovascular lesions, clinical events and symptoms in patients with familial CCM.
Methods
Primary endpoint is the new occurrence of clinical events CCM-related (intra-cerebral hemorrhage and focal neurological deficits). Secondary endpoints are (1) micro-vascular hemorrhages as assessed by magnetic susceptibility of the brain tissue, (2) cognitive function, global disability and health related quality of life, and (3) improvement of different vascular lesion characteristics. The trial is randomized, open-label, controlled, multicenter with central reading of MRI (PROBE design, Prospective Randomized Open Trial with Blinded Evaluation of Outcomes), and central adjudication of adverse events. Sample size calculations cannot be formally applied to the present Phase II trial, because the original number of eligible patients is expected not to be higher than 60. Special care will be paid to the biologic consistency of the different endpoints, even if none of them will yield statistically significant differences.
Results
Between April and December 2018, 37 patients (age 44 ± 14, female 51.4%) have been randomized in 5 Italian Clinical Centers. Baseline characteristics of the patients are reported in Table 1.
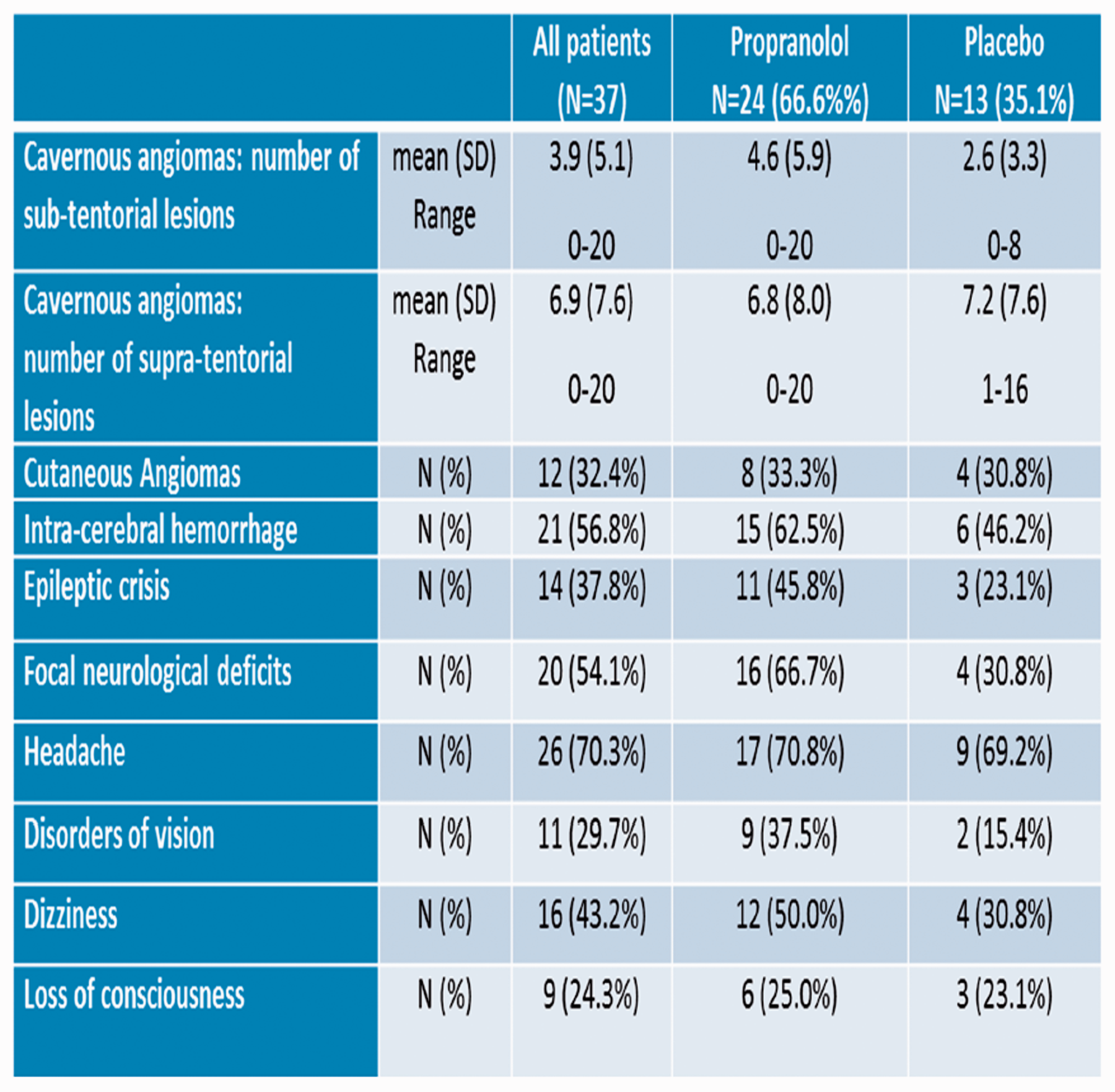
Conclusions
Last-patient-last-visit is scheduled Dec 2020. The possible extension of the trial, in case of encouraging preliminary results, is foreseen.
Trial registration number
ClinicalTrial.gov ID: NCT03589014
AS36-002
SYMPTOM AWARENESS AND TIME FORM ONSET TO INTERVENTION: THE “PRESTO” STUDY
1EO Galliera Hospital, Neurological Unit, Genova, Italy
2na, na, Genova, Italy
Background and Aims
Best medical treatments of ischemic stroke are admission to stroke unit, intravenous thrombolysis and, in selected cases, thrombectomy. Time from symptom onset to interventions is the best predictor of clinical outcome. Knowledge of stroke symptoms could reduce the time to intervention.
Methods
We planned a campaign in Genova, Italy, to spread the stroke symptoms knowledge. We registered time from onset to door (TOD) from february 1st 2018 to may 31st 2018 in the three Genova Hospitals. The campaign “PRESTO” started on June 1st 2018 and will finish at the end of January 2019; then, from February 2019 to June 2019 we will record again TOD after the campaign. The campaign consists in TV and radio spots, conference and visual explanation of the “PRESTO” message through the local media. PRESTO message is the translation of FAST (“Face, Arm, Speech, Time”) message in Italian language, with the adjunct of visive symptoms (amaurosis, diplopia) and headache.
Results
Until now, we recruited 470 patients, mean age 77.85 ± 12.29 years. Of them,370 (78.7%) arrived to emergency room (ER) calling the telephone Emergencies number (n.112), 72 (15.3%) self-presented and 28 (6%) through ambulance. TOD was 214 ± 251.84 minutes (range 4-1392 min). There was a statistically significant difference in TOD depending on modality of presentation to ER: self-presented vs. n.112 had a higher TOD with a difference of 105.9 minutes (p = .003, 95% CI = 28.7-183.1).
Conclusions
Calling 112 is the best way to reduce TOD. We will evaluate the effect of the campaign PRESTO on TOD at the end of the project.
Trial registration number
N/A
AS36-034
EEG CONTROLLED TRIAGE IN THE AMBULANCE FOR ACUTE ISCHEMIC STROKE TRIAL (ELECTRA-STROKE)
1Amsterdam University Medical Centers- location AMC, Department of Neurology, Amsterdam, The Netherlands
2Ambulance Amsterdam, Emergency Medical Services, Amsterdam, The Netherlands
Background and Aims
Endovascular thrombectomy (EVT) is the standard treatment for patients with acute ischemic stroke (AIS) and a large vessel occlusion (LVO). Direct presentation of patients with a suspected LVO in a comprehensive stroke center (CSC) reduces onset-to-treatment time by approximately an hour and thereby improves clinical outcome. However, a reliable tool for pre-hospital LVO-detection is currently not available. Preliminary electroencephalography (EEG) studies show that hemispheric hypoxia quickly results in slowing of EEG-signal. Dry electrode EEG caps allow reliable EEG measurement in less than five minutes. We hypothesize that dry electrode EEG is an accurate and feasible diagnostic test for LVO in the ambulance.
Methods
ELECTRA-STROKE is an ongoing diagnostic pilot study that consists of four phases. In the first two phases, optimization of EEG measurement time and software settings is achieved in a non-emergency setting. In the third phase, dry electrode EEGs are measured in 50 patients with a suspected AIS in the emergency room, and the data are used to select an algorithm for LVO-detection for phase four. In the final phase, paramedics will perform dry electrode EEGs in 222 patients with suspected AIS, to validate the algorithm and assess technical and logistical feasibility. The primary outcome is specificity of the LVO-detection algorithm and sample size calculation is based on an expected specificity of 70%.
Results
Study progress
This study started in October 2018. Phase 1 and 2 have been successfully completed. Phase 3 has commenced in December 2018 (currently 16 inclusions). Phase 4 is expected to start in May 2019.
Trial registration number
NCT03699397
AS36-003
APIXABAN FOR TREATMENT OF EMBOLIC STROKE OF UNDETERMINED SOURCE – ATTICUS RANDOMIZED TRIAL
1University Hospital Tuebingen, Neurology, Tuebingen, Germany
2University Hospital Tuebingen, Cardiology, Tuebingen, Germany
3University Hospital Tuebingen, Biometry, Tuebingen, Germany
4Katharinen Hospital, Neurology, Stuttgart, Germany
5Hospital Martha-Maria, Neurology, Halle-Döhlau, Germany
6Rems-Murr-Kliniken, Neurology, Winnenden, Germany
7Carl-von-Basedow Hospital, Neurology, Merseburg, Germany
8University Hospital Goettingen, Neurology, Goettingen, Germany
9University Hospital Goettingen, Cardiology, Goettingen, Germany
10Schwarzwald-Baar Hospital, Neurology, Villingen-Schwenningen, Germany
11Schwarzwald-Baar Hospital, Cardiology, Villingen-Schwenningen, Germany
12Hospital Friedrichshafen, Neurology, Friedrichshafen, Germany
13Marien Hospital, Neurology, Stuttgart, Germany
14Hospital Ludwigsburg, Neurology, Ludwigsburg, Germany
15Hospital Coburg, Cardiology, Coburg, Germany
16University Hospital Bonn, Neurology, Bonn, Germany
17University Hospital Kiel, Neurology, Kiel, Germany
18Medical Park Humboldtmühle, Neurology, Berlin, Germany
19University Hospital Ulm, Neurology, Ulm, Germany
20Johannes Wesling Hospital, Neurology, Minden, Germany
21University Hospital Magdeburg, Neurology, Magdeburg, Germany
22University Hospital Muenster, Neurology, Muenster, Germany
Background and Aims
Optimal prevention after embolic stroke of undetermined source (ESUS) is not established. ESUS is caused by thromboembolism and associated with high risk of recurrent stroke and clinically silent ischemic lesions (IL). Current standard in ESUS patients is acetylsalicylic acid (ASA), despite high prevalence of occult atrial fibrillation (AF).
ATTICUS will determine whether apixaban started ≤28 days after index stroke is superior to ASA for prevention of new IL on 12-months follow-up MRI in an AF-at-risk-enriched ESUS population, i.e. patients with dilated left atrium, reduced flow or spontaneous echo contrast in left atrial appendage on transesophageal echocardiogram, atrial runs on 24h-Holter-ECG, or an CHA2DS2-VASc ≥4.
Methods
Prospective, randomized (1:1), open, blinded-endpoint (PROBE), multicenter phase III trial aiming at enrolling ∼500 ESUS patients undergoing cardiac remote monitoring (mandatory).
Results
Event-driven trial with core-lab adjudicated primary outcome, i.e. occurrence of at least one new IL at 12 months vs baseline. Key secondary outcomes are the combination of recurrent ischemic/hemorrhagic strokes and systemic embolism, combination of MACE including recurrent stroke, myocardial infarction and cardiovascular death, combination of major and clinically relevant non-major bleeding, and change of cognitive function (Montreal Cognitive Assessment) and quality of life (EuroQol-5D, Stroke Impact Scale).
Conclusions
In contrast to the recently published or presented NAVIGATE and RESPECT ESUS, patients enrolled in ATTICUS need to exhibit additional AF-predicting factors. Furthermore, mandatory cardiac remote monitoring will help to elucidate the impact of AF and effects of early oral anticoagulation with apixaban compared to antiplatelet therapy with ASA on the incidence of new IL after ESUS.
Trial registration number
https://clinicaltrials.gov/ct2/show/NCT02427126
AS36-039
SECONDARY PREVENTION BY STRUCTURED SEMI-INTERACTIVE STROKE PREVENTION PACKAGE IN INDIA (SPRINT INDIA) STUDY
1Christian Medical College, Department of Neurology, Ludhiana, India
2Indian Council of Medical Research, Division of NCD, New Delhi, India
Background and Aims
Recurrent Stroke may occur in one-fifth of patients. Patient and Caregiver education of risk factors may help in stroke prevention. We hypothesise that a structured semi-interactive stroke prevention package may reduce the risk of recurrent strokes, myocardial infarction and death in patients with sub-acute stroke at the end of one year.
Methods
This is the first multi-centre randomised clinical stroke study involving 25 centres under INSTRuCT Network (Indian Stroke Clinical Trial Network). Patients with first ever sub-acute stroke within 2 days-3 months of onset, age 18-85 years, mRS<5, imaging evidence of stroke are included. Participants/caregivers able to read and complete tasks suggested in stroke workbook and have cellular device for receiving SMS (Short Message Service) and watching videos are eligible. A total of 5830 patients speaking 11 different languages are being randomised to intervention or control arm all over India. Patients in the intervention arm are receiving a stroke prevention workbook, regular educational short messages (108) and videos (17). The outcome event is a composite of adjudicated recurrent stroke, myocardial infarction or death over a period of one year.
Results
From May 2018 to 20 Jan 2019, 927 (16%) patients are screened and 735 (12.6%) are randomised. The total target of 10.5% is achieved. Intervention arm has 369 (50.2%) patients. Twelve events and 153 protocol deviations have been reported.
Conclusions
The result of study may bolster our efforts to implement patient and caregiver education strategies for stroke prevention.
Trial registration number
Clinical Trials.gov Identifier: NCT03228979
CTRI number: CTRI/2017/09/009600
AS36-041
ENDOLOW – ENDOVASCULAR THERAPY FOR LOW NIHSS ISCHEMIC STROKES
1University Hospital Heidelberg, Department of Neurology, Heidelberg, Germany
2University of Cincinnati, UC Stroke Team, Cincinnati, USA
3University of Calgary, Depts of Clinical Neurosciences and Radiology, Calgary, Canada
4Emory University School of Medicine, Marcus Stroke & Neuroscience Center, Atlanta, USA
5University of Heidelberg, Institute of Medical Biometry and Informatics, Heidelberg, Germany
6University of Cincinnati, Radiology, Cincinnati, USA
7Emory University School of Medicine, Department of Emergency Medicine, Atlanta, USA
8Emory University School of Medicine, Dept. of Emergency Medicine, Atlanta, USA
9University of Cincinnati, Division of Biostatistics and Epidemiology, Cincinnati, USA
Background and Aims
ENDOLOW is designed to determine the safety and efficacy of immediate mechanical thrombectomy (iMT) compared to immediate medical management (iMM) for patients with low National Instiute of Health Stroke Scales (NIHSS 0-5) and large vessel occlusion (LVOs).
Methods
ENDOLOW is a prospective, randomized, open-label, blinded-endpoint (PROBE), multi-center, multinational clinical trial. This investigator-initiated study is funded by Cerenovus, Inc. The study is an adaptive two-stage design allowing for early stopping for efficacy or futility at the interim, or continuation to stage two after reestimating the sample size. Included patients are over 18 years of age an NIHSS of 0-5 and presence of an objective neurologic deficit, LVO (intracranial ICA, M1, M2), ASPECTS ≥ 6, and a baseline mRS ≤ 2 who present within 24 hours (8-24h with perfusion mismatch) from last known well. The first stage will enroll up to 200 patients. Patients are randomized 1:1 to either iMT or iMM using a permuted block randomization stratified by IV tPA, time from onset to randomization, occlusion site, and age. The primary endpoint is the distribution of 90-day mRS (“shift”).
Results
ENDOLOW received formal confirmation of funding in September 2018. Regulatory requirements are being prepared. Participating sites from the United States, Canada and Europe are being identified. Update on trial progression will be presented at the time of the conference.
Conclusions
ENDO LOW is designed to establish the clinical value of iMT in the treatment of patients with LVOs and low NIHSS.
Trial registration number
pending …
AS36-042
SAFETY, PHARMACOKINETICS AND PHARMACODYNAMICS OF A NOVEL IMMUNOMODULATOR (APTOLL) AFTER INTRAVENOUS INJECTION IN HEALTHY VOLUNTEERS
1AptaTargets S.L., Science, Madrid, Spain
2Hospital Universitario La Princesa, Clinical Pharmacology, Madrid, Spain
3Thomas Rupp, Consulting, Berlin, Germany
4University Complutense, Pharmacology and Toxicology, Madrid, Spain
5Medical University of Vienna, Clinical Pharmacology, Vienna, Austria
6Aptus Biotech S.L., Investigation, Madird, Spain
7IRYCIS-Hospital Ramón y Cajal, Investigation, Madrid, Spain
Background and Aims
TLR4 receptor plays a key role activating inflammatory response that exacerbates brain damage after ischemia (Caso et al., Circulation2007). ApTOLL is an aptamer that blocks TLR4 activation in the acute phase, showing an outstanding profile reducing brain damage after ischemic stroke in rat and mouse models of permanent and transient experimental ischemia (Fernández et al., Mol Ther2018). In the reperfusion era, developing new neuroprotective drugs to ameliorate the reperfusion damage and further improve outcome in patients, is a very promising therapeutic strategy.
Methods
First-in-human dose-ascending, randomized, placebo-controlled phase I clinical trial to assess safety and pharmacokinetics (PK) of ApTOLL in 46 healthy adult male and female volunteers (HVs). The study is divided in two parts: part A (7 ascending dose levels) and part B (multiple dose with the maximum selected dose in the part A). ApTOLL is administered intravenously once in a slow infusion to each HV. Pharmacodynamics (PD) is also studied in an ex vivoLPS-challenge with blood from HVs from every dose level.
Results
Tolerability and adverse events (AEs) of all doses are assessed. Biochemistry, coagulation and complement activation are reported. PK parameters are also checked: exposure (Cmaxand AUC), mean half-life and accumulation after multiple dose. In the PD study, cytokine release (IL6, TNFα, CXCL10 and INFγ) from HVs blood cells after LPS stimulation is determined.
Conclusions
No results have been obtained at the moment, but preliminary data are expected before starting the ESOC conference.
Trial registration number
2018-001721-51
AS36-043
A HERBAL MEDICINE, GONGJINDAN, IN SUBJECTS WITH CHRONIC DIZZINESS: A PROSPECTIVE, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED CLINICAL TRIAL FOR EFFECTIVENESS, SAFETY, AND COST-EFFECTIVENESS
1Kyung Hee Korean Medice Hospital, Stroke center, Seoul, Republic of Korea
2Kyung Hee Korean Medice Hospital, Clinical Trial Center, Seoul, Republic of Korea
3Pusan National University, Korean Medical Ophthalmology & Otolaryngology & Dermatology, Busan, Republic of Korea
4Semyung University, Sasang Constitutional Medicine, Jecheon, Republic of Korea
5Dongguk University, Oriental Medicine Ophthalmology & Otolaryngology & Dermatology, Goyang, Republic of Korea
Background and Aims
This study aims to explore the effectiveness, safety, and cost-effectiveness of a herbal medication, Gongjindan (GJD), inpatients with chronic dizziness.
Methods
Seventy-eight patients diagnosed with Meniere’s disease, psychogenic dizziness, or dizziness of unknown cause will be randomized and allocated to either a GJD or a placebo group in a 1:1 ratio. Participants will be orally given 3.75 g GJD or placebo in pill form once a day for 56 days. The primary outcome is the Dizziness Handicap Inventory score. Secondary outcome measures are mean vertigo scale, visual analogue scale, frequency of dizziness, Berg Balance Scale, Fatigue Severity Scale, Qi Blood Yin Yang-deficiency questionnaire, Korean version of Beck’s Depression Inventory, and State-Trait Anxiety Inventory. To assess safety, adverse events will be monitored. Further, the incremental cost-effectiveness ratio will be calculated based on quality-adjusted life years and medical expenses.

Results
The primary and secondary variables will be statistically analyzed by analysis of covariance (ANCOVA) or rank ANCOVA with group and site as covariates. When there is a significant interaction between diagnosis (MD, psychogenic dizziness, or dizziness of unknown cause) and group, sub-group analysis will be performed. For safety assessment, the chi-squared test or Fisher’s exact test will be performed. Data will be statistically analyzed at a significance level of 0.05 (two-sided).
Conclusions
We are going to conduct a prospective, multicenter, randomized, double-blind, placebo-controlled, parallel-group, clinical trial on patients with chronic dizziness to evaluate the effectiveness, safety, and cost-effectiveness of a herbal drug, GJD.
Trial registration number
This trial is registered with ClinicalTrials.gov NCT03219515, in July 2017.
AS36-044
THE DUTCH ICH SURGERY TRIAL PILOT STUDY; MINIMALLY-INVASIVE ENDOSCOPY-GUIDED SURGERY FOR SPONTANEOUS INTRACEREBRAL HEMORRHAGE
1Donders Institute for Brain- Cognition and Behaviour- Radboud University Medical Center, Neurology, Nijmegen, The Netherlands
2Radboud University Medical Center- Nijmegen- The Netherlands, Neurosurgery, Nijmegen, The Netherlands
3Erasmus Medical Center, Neurology, Rotterdam, The Netherlands
4Elisabeth-Twee Steden Ziekenhuis, Neurology, Tilburg, The Netherlands
5Neurosurgical Center Amsterdam- Amsterdam University Medical Centers- VU University Medical Center and Academic Medical Center, Neurosurgery, Amsterdam, The Netherlands
6Leiden University Medical Center, Neurology, Leiden, The Netherlands
7University Medical Center, Neurology and Neurosurgery, Utrecht, The Netherlands
8Erasmus Medical Center- Erasmus MC Stroke Center, Neurosurgery, Rotterdam, The Netherlands
Background and Aims
Spontaneous intracerebral hemorrhage (ICH) is an important cause of global disease burden. ICH volume is an important predictor of outcome in patients with supratentorial ICH but reduction of ICH volume by craniotomy has failed to improve outcome. Increasing evidence suggests that minimally-invasive endoscopy-guided surgery reduces ICH volume and may improve functional outcome, especially when performed soon after symptom onset.
Methods
Multicenter, prospective, cohort study with blinded outcome assessment.
Study population
We will include patients with spontaneous supratentorial ICH. Forty patients in three participating centers will undergo minimally-invasive endoscopy-guided surgery within 8 hours of symptom onset. In seven other hospitals, we will include 360 consecutive patients treated with standard medical treatment for comparison of functional outcome.
Primary outcome
Primary safety outcome is a combination of death or neurological deterioration (increase in NIHSS with at least 4 points) within 24 hours. Primary technical outcome is the proportional volume reduction at 24 hours.
Secondary outcomes
Ordinal shift in modified Rankin Scale at 90 and 180 days.
Funding
Netherlands Cardiovascular Research Initiative, supported by the Dutch Heart Foundation, (CVON2015-01), Penumbra Inc.
Results
Since December 2018 11 patients have been included in the control arm (64% male, mean age 72) and since February 2019 we are waiting for the first patient in the surgical arm.
Conclusions
Status: Recruiting.
Trial registration number
NCT03608423
www.dutch-ich.nl
AS36-045
COLCHICINE FOR PREVENTION OF VASCULAR INFLAMMATION IN NON-CARDIOEMBOLIC STROKE (CONVINCE)
1University College Dublin, Stroke Clinical Trials Network Ireland, Dublin, Ireland
Background and Aims
Despite modern prevention treatments, the 5-year residual risk of recurrent stroke or coronary events is 25%. Increasing evidence suggests that inflammation plays a key role in stroke. In the CANTOS trial, canakinumab, an IL-1b antagonist, prevented recurrent vascular events in patients with coronary disease suggesting that anti-inflammatory therapy may have benefit after non-cardioembolic stroke. Low-dose colchicine is a potent, inexpensive anti-inflammatory therapy with an established safety profile
Methods
CONVINCE is an actively recruiting, prospective, open-label, blinded endpoint (PROBE) randomised trial comparing low-dose colchicine (0.5mg) plus usual care to usual care (lifestyle advice, antiplatelet, antihypertensive, lipid-lowering therapy) alone for the prevention of vascular events after non-severe non-cardioembolic stroke or high risk TIA. We aim to recruit 3100 patients in total over a 5 year period.
Inclusion criteria are age > 40 years with ischaemic stroke or high-risk TIA, with onset between 72 hours and 28 days, where the event mechanism is probably large-or small-artery disease, or cryptogenic stroke unlikely due to cardio-embolism or other specified causes.
The primary end-point is time to non-fatal or fatal recurrent ischemic stroke or acute coronary events.
Results
Currently, 93 sites are enrolling across Belgium, Czech Republic, Germany, Ireland, Poland, Spain, and UK. 770 patients have been randomised. Sites in Estonia, Lithuania, Denmark, Portugal, Canada, UAE, Israel, Netherlands, Switzerland, Italy, and North Macedonia will open in 2019. Updated site details and recruitment figures will be presented at the European Stroke Conference
Conclusions
The CONVINCE trial is testing anti-inflammatory therapy with low-dose colchicine plus guideline-based usual care for prevention of recurrent stroke and major vascular events compared with usual care alone.
Trial registration number
NCT02898610
AS36-046
THE PARAMEDIC NORWEGIAN ACUTE STROKE PREHOSPITAL PROJECT DIAGNOSTICS AND TRIAGE OF ACUTE STROKE BY PARAMEDICS USING THE NATIONAL INSTITUTE OF HEALTH STROKE SCALE (NIHSS)
1Norwegian Air Ambulance Foundation, Research department, Oslo, Norway
2Oslo University Hospital, Department of Neurology, Oslo, Norway
3Oslo University Hospital, Division of Prehospital Services, Oslo, Norway
4University of Oslo, Institute of Clinical Medicine, Oslo, Norway
Background and Aims
Prehospital recognition of stroke symptoms is the first step in the stroke treatment chain and therefore imperative for reducing time to treatment. Today paramedics in Norway use FAST for recognizing stroke patients, which is a rapid examination of a suspected stroke patient but lacks the sensitivity and specificity of the NIHSS. In the ParaNASPP study we will investigate the use of NIHSS by paramedics in a prehospital setting with the aid of a mobile application, the eSTROKE app.
Methods
In this randomized stepped wedge cluster design study, we aim to include 1300 patients with suspected stroke prehospitally. The trial will include five ambulance stations that service the Oslo University Hospital. The intervention is prehospital triage of stroke patients by paramedics specially trained in NIHSS and the eSTROKE app. The eSTROKE app will include NIHSS score, age, time of onset, list of anticoagulant medication and vital parameters. Recorded data in the eSTROKE app is sent directly to the on-call stroke physician, in addition to telephone communication. The primary outcome is the positive predictive value (PPV) of a final stroke related diagnosis. Secondary outcomes include time variables, prehospital NIHSS quantification, infarction volume and functional outcome at 90 days.
Results
The implementation of prehospital NIHSS with the eSTROKEapp will make paramedics more adept at examining suspected stroke patients, enable more fluent communication with in-hospital services and therefore facilitate more correct triage and transportation to the right level of care.
Conclusions
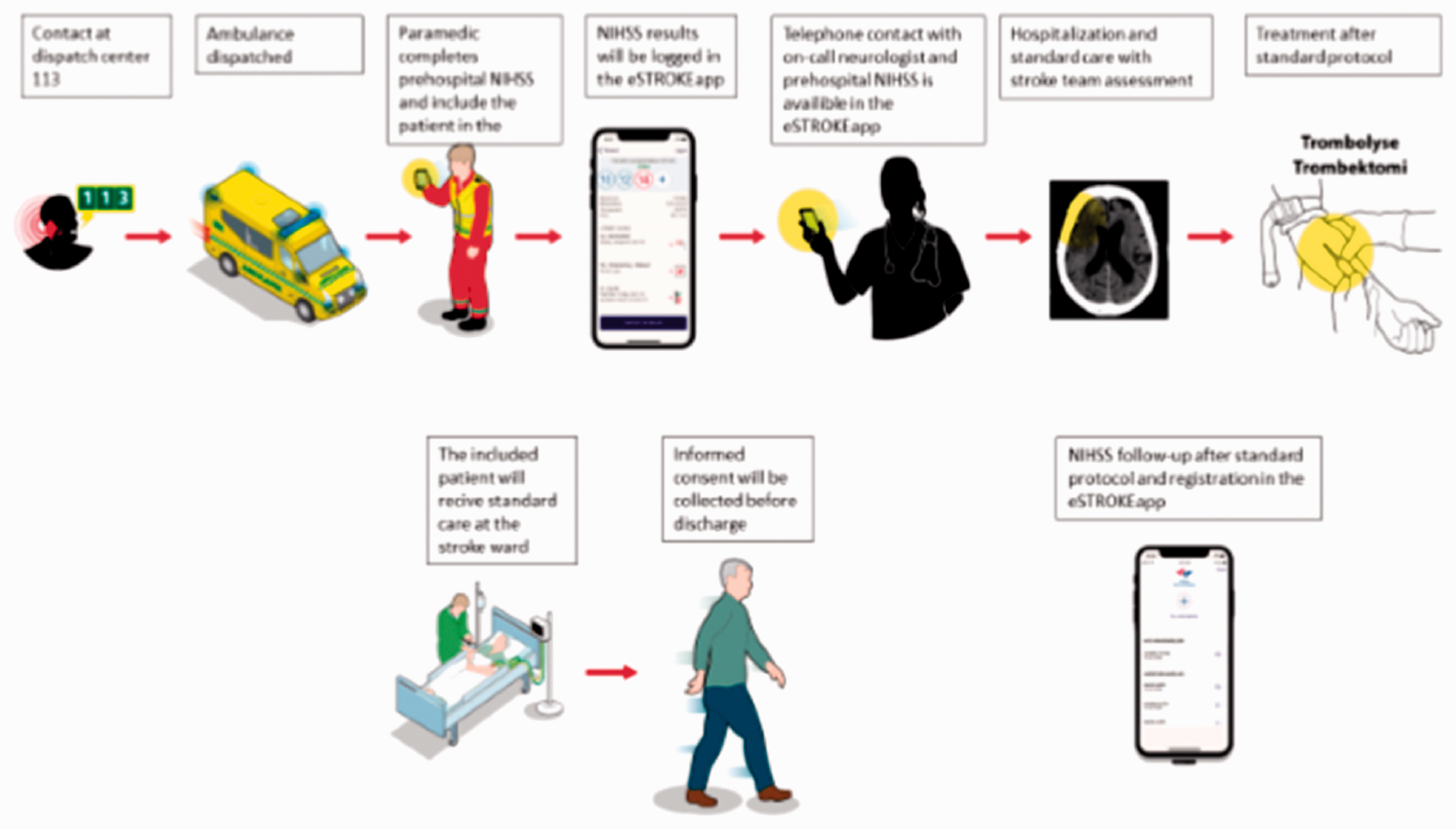
Trial registration number
REK2018/2310
AS36-047
SCREENING AND PATIENT-TAILORED CARE FOR EMOTIONAL AND COGNITIVE COMPLAINTS IN PATIENTS DISCHARGED HOME AFTER ISCHEMIC STROKE (ECO-STROKE TRIAL)
1OLVG Amsterdam, Neurology, Amsterdam, The Netherlands
2Maastricht University, Clinical Neuropsychology, Maastricht, The Netherlands
3UMC Utrecht, Rehabilitation, Utrecht, The Netherlands
Background and Aims
Most patients diagnosed with minor ischemic stroke are likely to be discharged home. At home, a substantial number of these patients experience cognitive and emotional complaints, resulting in decreased quality of life and decreased societal participation. Therefore, this study examines the effect of screening and patient-tailored care for cognitive and emotional complaints in patients discharged home after ischemic stroke.
Methods
In a multicenter, single blinded, cluster randomized controlled trial, 12 hospitals will be randomized (1:1) to the intervention group or the usual care group. Patients discharged home after ischemic stroke without rehabilitation care will be included. The intervention encompasses one consultation of 60 minutes executed by a specialized nurse at the outpatient clinics 6 weeks after the diagnosis. This consultation includes 1) screening for cognitive and emotional problems using sensitive instruments, 2) active information provision and decision making according to the principles of self-management support and 3) a protocol for referral if needed.
Results
The primary outcome will be the level of participation measured with the Restriction subscale of the Utrecht Scale for Evaluation of Rehabilitation-Participation at 1 year. Secondary outcomes will be the level of cognitive complaints, depression and anxiety, quality of life, patient-reported global health and physical disability at 1 year. Additionally, a cost-analysis will be performed. Finally, a mixed-methods study will assess the feasibility.
Conclusions
The combination of clinical and cost-effectiveness and feasibility will have to demonstrate the added value of this intervention over care as usual for ischemic stroke patients.
Trial registration number
NTR7504
AS36-048
EMOTIONAL AND COGNITIVE DETERMINANTS OF LONG-TERM FATIGUE AFTER STROKE. A PROSPECTIVE STUDY
1The University Hospital of North Norway, Department of Rehabilitation, Tromsø, Norway
2The University Hospital of North Norway, Department of Paediatric Rehabilitation, Tromsø, Norway
3The University Hospital of North Norway- Heart and Lung Clinic, Department of Cardiothoracic and Vascular Surgery, Tromsø, Norway
4University of Tromsø – Arctic University of Norway- Faculty of Health Sciences, Department of Health and Care Sciences, Tromsø, Norway
5University of Tromsø – Arctic University of Norway- Faculty of Health Sciences, Department of Clinical Medicine, Tromsø, Norway
6University of Tromsø – Arctic University of Norway- Faculty of Health Sciences, Department of Psychology, Tromsø, Norway
7University of Oslo- Institute for Health and Society, Department of Nursing Science, Oslo, Norway
8Regionshospital Hammel Neurocenter, Department of Clinical Medicine, Aarhus, Denmark
9University of Tromsø – Arctic University of Norway- Faculty of Health Sciences, Department of Clinical Medicine, Tromsø, Norway
Background and Aims
Persistent fatigue is a frequent sequela in adult stroke survivors, but knowledge concerning underlying psychological mechanisms is scarce. Since one of four stroke patients perceive fatigue as one of the most difficult symptoms to cope with, this problem is important to address. The objectives of this study are to examine fatigue four years after stroke, as well as associations with cognitive functioning, repeated measures of anxiety and depression, metacognitions and health-related quality of life.
Methods
Study design: Prospective observational study.
Participants: Adults with acute ischaemic or haemorrhagic stroke admitted to the three stroke units at the University Hospital of North Norway during 2014 and 2015. Four years after stroke, follow-up questionnaires are administrated to all who completes questionnaires at 3 and 12 months after stroke. Patients below 75 years of age are invited to a neuropsychological assessment, including a one-week recording of physical activity and sleep patterns.
Exclusion criteria: Injuries or diseases that prevent a valid assessment.
Primary outcome: Fatigue as measured with the Fatigue Severity Scale and the Chalder Fatigue Questionnaire.
Other measures: Neuropsychological assessments of cognitive function. Self-report measures include Stroke Specific Quality of Life Scale, Hospital Anxiety and Depression Scale, and Metacognitions Questionnaire. Physical activity and sleep pattern measured with accelerometer. Baseline registrations from the National Norwegian Stroke Registry (previous stroke, stroke subtype, lesion location, comorbidity, degree of disability measured with the Modified Rankin Scale).
Results
Recruitment of 150 participants will be completed in May 30, 2019.
Conclusions
Main results will be available approximately 2020-2021. Estimated completion date: December 2021.
Trial registration number
NCT03639259.
AS36-049
THE EFFECT OF A TABLET-BASED APHASIA THERAPY IN THE CHRONIC PHASE AFTER STROKE
1University Ghent Hospital, Neurology, Ghent, Belgium
2University Ghent Hospital, Otorhinolaryngology, Ghent, Belgium
3University Ghent Hospital, Physical Medicine and Rehabilitation, Ghent, Belgium
4University Ghent, Rehabilitation Sciences, Ghent, Belgium
5University Medical Centre Groningen, Centrum for Rehabilitation, Groningen, The Netherlands
Background and Aims
Aphasia after stroke can majorly impact a persons’ quality of life. Intensive aphasia therapy can improve language functions, even in the chronic stage of recovery. However, the desired intensities for language rehabilitation are often challenging to achieve due to limited health care resources, transportation difficulties, fatigue etc. New technological developments such as computer- and tablet-based aphasia therapies as an add-on to conventional face-to-face therapy might be a promising rehabilitation tool to enhance therapy effects.
This study will investigate the clinical, functional and neurophysiological effects of intensive tablet-based aphasia therapy as an add-on to conventional face-to-face therapy compared to conventional therapy alone in patients with aphasia following stroke.
Methods
A randomized, parallel-group, sham-controlled, single-blinded clinical trial will be performed. Thirty-six right-handed persons with aphasia following a first left hemispheric stroke in the chronic stage of recovery ( > 6 months post-stroke) will be randomly allocated into one of three groups. After repeated baseline measurements, all groups will receive face-to-face therapy for 3 weeks (3hours/week). In the active condition, patients will practice an additional 5 hours/week with a language app ‘STAPP’, whereas a first sham group will use a tablet in a recreational way and a second sham group will be restricted from tablet use. Therapy-effectiveness will be measured by specific linguistic tests, functional communication abilities, quality of life and event-related potentialsat baseline, immediately before and after intervention, and at 3 months follow-up.
Results
Data collection is ongoing.
Conclusions
We hope to confirm that additional tablet-based aphasia therapy can improve functional outcome.
Trial registration number
Clinicaltrials.gov: NCT03622411, registered since 08/06/2018
AS36-050
EFFECTS OF AN AVOCADO BASED MEDITERRANEAN DIET ON SERUM LIPIDS FOR SECONDARY PREVENTION AFTER ISCHEMIC STROKE TRIAL (ADD-SPISE)
1Clínica Alemana de Santiago, Neurology and Psychiatry, Santiago, Chile
2Universidad del Desarrollo, Centro de Química Médica, Santiago, Chile
Background and Aims
Dietary risk has a high impact in DALYs in acute ischemic stroke (AIS). Adherence to dietary patterns like the Mediterranean Diet (MeDi) are associated with a decreased risk of stroke mortality/incidence in observational studies and primary prevention trials. Furthermore, lowering Low Density Cholesterol (LDL-C) levels decreases stroke recurrence. Interestingly, Avocado-substituted diets significantly decrease cholesterol levels. Aim: To determine the effect of a MeDi based on Avocados on lipid profile after an AIS.
Methods
Methods and patients: Academic, open-label, blinded outcome assessment, controlled clinical trial. Recruitment within a month of AIS if fulfilling eligibility criteria (including consent) and randomly assigned to the diet intervention in a 1:1 ratio. A) Avocado based MeDi with daily intake of ½ an avocado B) Low fat-high complex carbohydrate healthy-diet. Study outcomes: Main outcome: Level of plasma LDL-C at 3 months of intervention. Secondary outcomes: Levels of lipid profile, inflammation markers, glycemic control, anthropometric measures, stroke recurrence, cardiovascular events, adverse events, compliance.
Sample size estimates: A total sample of 200 patients was estimated to provide 80% power and 5% level of significance (10% loss and 5% crossover) to detect a 4.6 ml/dL difference of in LDL-C.
The study has ethics committee approval.
Results
The trial is progressing as planned. From August 2018 to February 2019, 25 patients have been recruited and 15 have completed 3-month follow-up.
Conclusion
We hypothesize that an Avocado based MeDi will significantly reduce levels of LDL-cholesterol at 3 months in patients who have suffered an AIS compared to the control diet.
Trial registration number
ADDSPISE Clinical Trials.gov Identifier NCT03524742.
AS36-051
THE USE OF OUTCOME DATA IN SHARED DECISION MAKING FOR STROKE PATIENTS
1OLVG, Neurology, Amsterdam, The Netherlands
2OLVG, Quality and improvement, Amsterdam, The Netherlands
3Medisch Spectrum Twente, Neurology, Enschede, The Netherlands
4Amsterdam UMC location AMC, Neurology, Amsterdam, The Netherlands
Background and Aims
The Santeon network consists of seven leading teaching hospitals in the Netherlands that altogether account for 11% of the nation’s hospital care volume. Value based health care is a fundamental element of Santeon’s strategy. In accordance with the value based health care principles, individual stroke care could be optimized by promoting shared decision making. However, best practices regarding the actual implementation of shared decision making in stroke care are limited.
Methods
The first phase of this study will focus on improving shared decision making by facilitating decision support for stroke patients and their relatives. Potential choices will be identified, including rehabilitation options, healthcare professionals will be trained, and an implementation plan will be constructed to embed the shared decision making process within the existing care pathways. Action research and a pre-post study is planned to evaluate the effects on patients’ satisfaction, empowerment and treatment choices. The second phase of this study comprises the development of a “patients’ like me” or clinical prediction model for stroke patients based on several outcome data, including functional- and patient-reported outcome measures. The third part of this study covers the implementation of both the decision aid and the clinical prediction model in all Santeon hospitals in the Netherlands.
Results
This project will determine a more individualized approach both at admission and discharge, based on the needs and preferences of stroke patients and their relatives, aiming to increase patients’ satisfaction, enhance their quality of life, promote relevant care, and reduce healthcare costs.
Conclusions
Will be obtained.
Trial registration number
N/A
WITHDRAWN
AS36-053
AVAILABILITY AND SAFETY OF SEQUENTIAL PROSTAGLANDIN E1 TREATMENT IN CEREBRAL SMALL VESSEL DISEASE
1Drum Tower Hospital- Nanjing University Medical School, Department of Neurology, NanJing, China
Background and Aims
Alprostadils are potent vasoactive agents with wide variety of other actions such as vasodilatation, fibrinolysis and inhibition of platelet aggregation, which coincide with several potential pathophysiology mechanism of cerebral small vessel disease (CSVD). Prostaglandin E1 (PGE1) is the most widely used pharmaceutic among all the alprostadils related drugs. This ongoing trial aims to evaluate the efficacy and safety of sequential PGE1 treatment in patients with CSVD. Hypothesis: After the sequential PGE1 treatment in CSVD, the safety results in the treatment group would be comparable with the control group. Besides, the patients in treatment group would obtain benefit in cognition.
Methods
See supplementary picture.

Results
After 1-year, 119 subjects (PGE1 treatment group, n = 59, control group, n = 60) in all were included at present and completed the follow-up. At the end of follow-up, the risk of ischemic stroke was statistically significantly lower in the PGE1+aspirin group than in the aspirin group (relative risk, 0.74 [95% CI, 0.65 to 0.88]). In contrast, the risk for hemorrhagic events was similar between the 2 groups (relative risk, 0.92 [CI, 0.79 to 1.13]). The PGE1+aspirin group displayed a slower rate of decline in MoCA scores (P = 0.047). Lower WMH progression and less number of lacunar infarction were shown in the PGE1+aspirin group (P = 0.022 and P = 0.045 respectively).
Conclusions
According to the current results of the 119 subject, PGE1 reduced the risk of ischemic stroke recurrence and retard the progression of cognitive decline and WMH progression in CSVD.
Trial registration number
Clinical registration number: ChiCTR-INR-17011209
AS36-054
THE NIH STROKENET TRIALS IMPLEMENTATION
1University of Cincinnati, Neurology and Rehabilitation Medicine, Cincinnati, USA
2Medical University of South Carolina, Public Health Sciences, Charleston, USA
3The National Institutes of Health, The National Institute of Neurological Disorders & Stroke, Bethesda, USA
Background and Aims
The NIH StrokeNet infrastructure, established in 2013, consists of the National Coordinating Center at the University of Cincinnati that provides coordination of network activities; the National Data Management Center at the Medical University of South Carolina that coordinates centralized and standardized data collection and provides statistical support; 29 Regional Coordinating Centers that manage over 400 network sites; and the NINDS team that provides administrative and scientific input. The StrokeNet uses the Central Institutional Review Board, and has a Training and Education Core for the mentoring of StrokeNet fellows.
Methods
In the earlier years of the network, the StrokeNet’s evolving trial submission and review process had been a high priority of network development. Attention is now more focused on implementing the trials that have been developed and funded through that review process.
Results
Of 28 unique trial proposals submitted for NIH peer review, 12 have been approved for funding (6 prevention, 3 acute, and 3 recovery trials). The first StrokeNet trial approved for funding, DEFUSE 3, recruited ahead of projection and was halted early by the DSMB for overwhelming efficacy. TeleRehabilitation met recruitment projections and finished on schedule. ARCADIA is currently activating 150 sites and enrolling patients. MOST, Sleep SMART, TRANSPORT2, and I-ACQUIRE trials are funded and will be implemented by summer of 2019. ARCADIA-CSI, SATURN and ASPIRE are expected to commence enrollment by fall of 2019 and FASTEST expects to start by beginning of 2020.
Conclusions
The NIH StrokeNet has demonstrated the ability to design and implement innovative and impactful stroke trials.
Trial registration number
N/A
AS36-055
OPTIMAS: A RANDOMISED CONTROLLED TRIAL TO ESTABLISH THE OPTIMAL TIMING OF ANTICOAGULATION AFTER ACUTE ISCHAEMIC STROKE
1University College London, Stroke Research Centre, London, United Kingdom
2University College London, Comprehensive Clinical Trials Unit, London, United Kingdom
3University College London, Research Department of Haematology, London, United Kingdom
4Royal Devon and Exeter NHS Foundation Trust, Stroke Department, Exeter, United Kingdom
5University of Liverpool, Liverpool Centre for Cardiovascular Science, Liverpool, United Kingdom
6University of Nottingham, Faculty of Medicine and Health Sciences, Nottingham, United Kingdom
Background and Aims
Anticoagulation is highly-effective for long-term stroke prevention in AF, but its safety and benefit in acute stroke is unclear. Current practice, based on expert opinion and guidelines, is to delay anticoagulation by up to two weeks, during which the risk of recurrent ischaemic stroke is 0.5%/day. Although early anticoagulation with warfarin or heparin increases the risk of symptomatic intracranial haemorrhage (sICH), observational evidence suggests that the early use of direct oral anticoagulants (DOACs), which pose lower bleeding risk, may be safe, offering the opportunity to reduce early ischaemic stroke risk without increasing ICH risk. OPTIMAS will test this hypothesis.
Methods
OPTIMAS will enrol 3478 participants with acute ischaemic stroke and AF in whom there is uncertainty regarding the optimal timing of anticoagulation. We will recruit over three years from April 2019 from > 100 stroke services in the United Kingdom. Participants will be randomised 1:1 to early (within 4 days) or standard (between day 7 and 14 after stroke) initiation of anticoagulation with any licensed DOAC.
Results
Outcomes will be assessed 90 days from randomisation, blinded to treatment allocation. The primary outcome is the combined incidence of recurrent ischaemic stroke, sICH and systemic embolism. A gatekeeping non-inferiority statistical design will be used. Secondary outcomes include bleeding, functional, cognitive, quality-of-life, and health-economic measures.
Conclusions
OPTIMAS will determine the efficacy and safety of early anticoagulation with a DOAC, a strategy with the potential to prevent early recurrent ischaemic strokes, reduce hospital stays, and change acute stroke care pathways for patients with AF.
Trial registration number
NCT03759938
ISRCTN17896007
AS36-056
PROGRESS OF DIRECT INTRA-ARTERIAL THROMBECTOMY IN ORDER TO REVASCULARIZE AIS PATIENTS WITH LARGE VESSEL OCCLUSION EFFICIENTLY IN CHINESE TERTIARY HOSPITALS: A MULTICENTER RANDOMIZED CLINICAL TRIAL
1Changhai Hospital- Naval Medical University, Neurosurgery, Shanghai, China
2Changhai Hospital- Naval Medical University, Neurology, Shanghai, China
3Amsterdam University Medical Centers, Radiology & Nuclear Medicine, Amsterdam, The Netherlands
4Amsterdam University Medical Centers, Neurology, Amsterdam, The Netherlands
Background and Aims
Intravenous thrombolysis (IVT) combined with mechanical thrombectomy (MT) has been proven safe and effective in patients with acute ischemic stroke (AIS) of anterior circulation large vessel occlusion (LVO). The incidence of symptomatic intracerebral hemorrhage was similar between bridging therapy and IVT, suggesting that this complication could not be attributed to the MT, but rather to IVT. Meanwhile, the incidence of intracranial atherosclerosis stenosis (ICAS) is higher in Asians. The primary objective is to assess the effect of MT compared to bridging therapy in patients with AIS due to anterior circulation LVO. The secondary objective is to assess treatment effect modification by presence of ICAS.
Methods
This is a parallel group randomized clinical trials of direct MT versus bridging therapy, using a non-inferiority design. The trial has observer blinded assessment of clinical outcome and baseline and follow-up neuro-imaging. The trial is executed in collaboration with MR CLEAN-NO IV investigators. Patients with anterior circulation AIS and LVO confirmed by CTA. Initiation of IVT must be feasible within 4.5 hours from symptom onset. Age must be 18 or over and NIHSS 2 or more. The trial aims to randomize 636 patients 1:1 to direct MT or bridging therapy as usual. The main outcomes is the score on the modified Rankin Scale at 3 months.
Results
The trial is currently running in 42 centers in the China. Patient inclusion started in February 2018. Until Marth 20 2019, the total number of enrolled cases was 458. Enrollment is expected to be completed in August, 2019.
Conclusions
N/A
Trial registration number
ClinicalTrials.gov Identifier: NCT03469206
AS36-057
STUDY OF ANTITHROMBOTIC TREATMENT AFTER INTRACEREBRAL HAEMORRHAGE (STATICH)
1Oslo University Hospital, Deptartment of Internal Medicine, Oslo, Norway
2University of Oslo, Institute of Clinical Medicine, Oslo, Norway
3Akershus University Hospital, Department of Neurology, Lørenskog, Norway
4Umeå University Hospital, Department of Public Health and Clinical Medicine, Umeå, Sweden
5Herlev Gentofte Hospital and University of Copenhagen, Department of Neurology and Neurovascular research unit, Herlev, Denmark
6Karolinska Institute- Danderyds Hospital, Department of Clinical Sciences, Stockholm, Sweden
7Nordland Hospital Trust, Department of Neurology, Bodø, Norway
8UiT The Arctic University of Norway, Department of Clinical Medicine, Bodø, Norway
Background and Aims
Patients with prior intracerebral haemorrhage (ICH) often have indication for antithrombotic treatment (antiplatelet or anticoagulant agents) for prevention of ischaemic events, but the benefit of such treatment is uncertain. STATICH will assess effects of antithrombotic treatment on the risk of recurrent ICH and ischaemic events after ICH, and examine the importance of MRI findings for these effects.
Methods
STATICH is a Scandinavian, multicentre, randomised controlled, open trial of antithrombotic treatment in patients with primary ICH during the last six months who have indication for antithrombotic treatment. Participants with vascular disease and indication for antiplatelet treatment are allocated to antiplatelet treatment or no antithrombotic treatment. Participants with atrial fibrillation and indication for anticoagulant treatment are allocated to anticoagulant or no anticoagulant treatment. Cerebral CT and MRI are performed before randomisation. Duration of follow-up is at least two years. The primary outcome variable is recurrent ICH. Important secondary outcome variables include ischaemic events and death. Assessment of clinical outcomes is performed blinded to treatment allocation. Target recruitment is 500 participants.
Results
Recruitment is on-going, and interested centres are welcome to participate. As per March 2019, more than 20 centres have been initiated, and 10 participants enrolled. The primary analysis will assess effects of antithrombotic treatment on clinical events. Secondary analyses will examine the importance of MRI findings like cerebral microbleeds for these effects.
Conclusions
Randomised controlled trials like STATICH are needed to evaluate the net effect of antithrombotic treatment after ICH. An individual patient data meta-analysis is planned with similar on-going trials.
Trial registration number
EudraCT number 2014-002636-13.
AS36-058
A MULTIMODAL INDIVIDUALIZED INTERVENTION TO PREVENT FUNCTIONAL DECLINE AFTER STROKE. A RANDOMISED CONTROLLED TRIAL ON LONG-TERM FOLLOW-UP AFTER STROKE (THE LAST-LONG TRIAL)
1Norwegian University of Science and Technology, Department of Neuromedicine and Movement Science, Trondheim, Norway
2Trondheim University Hospital, Department of Geriatrics, Trondheim, Norway
3Trondheim University Hospital, Department of Stroke, Trondheim, Norway
4Akershus University Hospital, Neurology, Lillestrøm, Norway
5Trondheim kommune, Rådmannens fagstab, Trondheim, Norway
Background and Aims
Physical and cognitive impairments are barriers to maintain function after stroke. The main objective of the LAST-long trial is to investigate the benefit of regular follow-up by a stroke-coordinator who sets up a treatment plan targeting the individual needs for follow-up in order to prevent functional decline in the long term after stroke.
Methods
A pragmatic randomised controlled trial, with repeated measures at 6, 12 and 18 months after inclusion, will be applied. Patients admitted to two Norwegian university hospitals will be screened for inclusion 3-month after stroke. A total of 490 patients, age ≥18, mRS <5, able to understand Norwegian and willing to sign informed consent, will be included. Patients with short life expectancy or other serious diseases will be excluded.
The intervention consist of regular meetings with a community-based stroke-coordinator, who will assess the patients’ risk-profil within physical health and lifestyle, mobility and ADL function, cognitive function and social function. For patients at risk within one or more of the domains, the coordinator will set up an action plan aiming to maintain or improve function. Only, already available community based or hospital based interventions will be accessed.
Mixed models will be used to evaluate differences between the groups for the primary (modified Rankin Scale) and secondary endpoints (cognition, motor function, extended ADL, self-perceived health, frailty, vascular events, caregivers burden, health costs, etc.) across the 4 time points.
Results
The first patient will be included in April 2019.
Conclusions
The LAST-long trial will be continued until 2023.
Trial registration number
ClinicalTrials.gov identifier: NCT03859063
AS36-059
TRANSDURAL REVASCULARIZATION ON MULTIPLE BURRHOLE WITH SYSTEMIC ERYTHROPOIETIN IN PATIENTS WITH CEREBRAL PERFUSION IMPAIRMENT
1Ajou university school of medicine, Department of Neurology, Suwon, Republic of Korea
2Ajou university school of medicine, Department of Neurosurgery, Suwon, Republic of Korea
Background and Aims
The revascularization effect of multi-burrhole and systemic erythropoietin (EPO) for neovascularization is still uncertain in symptomatic stroke patients with perfusion impairment. Therefore, we aimed to compare the transdural revascularization efficacy of the combination therapy group and burrhole-only groups.
Methods
This is a prospective, randomized, placebo-controlled, blinded-endpoint drug trial interim analysis that enrolled adult ischemic stroke patients who had perfusion impairment of grade ≥2 of within 7 days, atherosclerotic or steno-occlusive mechanism on CTA or MRA, no transdural collaterals by cerebral angiography. Patients were randomly assigned to multi-burrhole under local anesthesia plus EPO or to multi-burrhole alone. A total of 44 patients are the final recruitment number. Primary outcomes were revascularization success (trans-hemispheric, trans-burrhole, and relative revascularization area) at 6 months. Treatment-related adverse events were analyzed as secondary outcome.
Results
Thirty-seven hemispheres from 37 patients were included. There was no difference on baseline characteristics between the groups. As compared to multi-burrhole only group, successful revascularization was more prevalent in the combined treatment group: trans-hemispheric arteriogenesis (n = 17,94.4% vs. n = 10,52.6%; p = 0.008), trans-burrhole arteriogenesis (n = 36,73.5% vs. n = 24,46.2%; p = 0.005), and relative revascularization area (n = 6,33.3% vs. n = 3,15.8% in excellent, n = 3,16.7% vs. n = 5,26.3% in good, n = 8,44.4% vs. n = 2,10.5% in fair, n = 1,5.6% vs. n = 9,47.4% in poor; p = 0.005). There was no periprocedural (<2 weeks) cerebral infarction. After adjustment of potential factors, presence of moyamoya vessels (16.89, 1.83-156.28;p = 0.013) was an independent predictor for sufficient revascularization (filling ≥33% of ipsilateral supratentorium).
Conclusions
Combination therapy is feasible and safe in acute steno-occlusive patients with perfusion impairment. Successful revascularization appears to be associated with the presence of moyamoya vessels.
Trial registration number
ClinicalTrials.gov:NCT02603406
AS36-060
EARLY VERSUS LATE INITIATION OF DIRECT ORAL ANTICOAGULANTS IN POST-ISCHAEMIC STROKE PATIENTS WITH ATRIAL FIBRILLATION (ELAN): AN INTERNATIONAL MULTICENTRE, RANDOMISED-CONTROLLED, TWO-ARM, ASSESSOR-BLINDED TRIAL
1Inselspital, Department of Neurology, Bern, Switzerland
2University Hospital Helsinki, Department of Neurology, Helsinki, Finland
3University Hospital Perugia, Department of Internal Medicine, Perugia, Italy
4University Clinics Hamburg-Eppendorf, Department of Neurology, Hamburg, Germany
5University of Thessaly, Department of Internal Medicine, Larissa, Greece
6University Hospital Basel, Department of Neurology, Basel, Switzerland
7Cantonal Hospital Aarau, Department of Neurology, Aarau, Switzerland
8University Hospital Lausanne, Department of Neurology, Lausanne, Switzerland
9University Hospital and University of Bern-, Clinical Trial Unit, Bern, Switzerland
10University Clinics Graz, Department of Neurology, Graz, Switzerland
11Oslo University Hospital, Department of Neurology, Oslo, Norway
12Bern University Hospital, Department of Neurology, Bern, Switzerland
13University of Glasgow, Department of Neurology, Glasgow, United Kingdom
Background and Aims
When to start anticoagulation in ischaemic stroke patients with atrial fibrillation (AF) is a relevant unanswered question. Direct oral anticoagulants (DOACs) are at least as effective as Vitamin K antagonists in reducing stroke and systemic embolism and have a lower risk of intracranial haemorrhage. The ELAN trial will determine the net benefit of early versus late initiation of DOACs in patients with acute ischaemic stroke related to AF with respect to stroke size.
Methods
This international, multicentre, randomised-controlled, two-arm trial will randomise 2’000 patients into the experimental arm (early treatment; 1’000) or control arm (late treatment, 1’000). The time of DOAC initiation will vary depending on stroke size (i.e., minor, moderate, or major). Main inclusion criteria are signed informed consent, age > 18 years, confirmed ischaemic stroke and AF and agreement of treating physician to prescribe DOACs. The primary outcome is a composite of major bleeding, recurrent ischaemic stroke, systemic embolism and/or vascular death at 30 ± 3 days after randomisation.
Results
Recruitment at 13 sites in Switzerland and 16 sites in EU has started end of 2017. Approximately 80 sites in 10 European countries will eventually be recruiting patients.
Conclusions
This pragmatic trial will add evidence to the best time of starting DOACs after ischaemic stroke in patients with AF. If earlier initiation of DOACs in patients with ischaemic stroke related to AF is shown to be safe and efficacious, this could have a major impact on better treatment adherence, length of hospital stay and patient outcome. ClinicalTrials.gov Identifier: NCT03148457
Trial registration number
ClinicalTrials.gov Identifier: NCT03148457
AS36-061
SWISS TRIAL OF INITIAL DECOMPRESSIVE CRANIECTOMY VERSUS BEST MEDICAL TREATMENT OF SPONTANEOUS SUPRATENTORIAL INTRACEREBRAL HEMORRHAGE: A RANDOMIZED TRIAL (SWITCH)
1Inselspital, Department of Neurosurgery, Bern, Switzerland
2Helsinky University Central Hospital, Department of Neurology, Helsinki, Finland
3Johannes Gutenberg University Mainz, Department of Neurosurgery, Mainz, Germany
4Inselspital, Department of Neurology, Bern, Switzerland
Background and Aims
Decompressive craniectomy (DC) is beneficial in patients with various diseases including malignant middle cerebral artery (MCA) infarction. In intracerebral hemorrhage (ICH), decompressive craniectomy without hematoma evacuation has only been evaluated in small retrospective studies with a trend towards reduced mortality. However, no randomized trial has ever assessed whether DC is beneficial in ICH. Therefore, the prospective randomized SWITCH trial was initiated in October 2014 to determine whether decompressive surgery and best medical treatment in patients with spontaneous ICH will improve outcome compared to best medical treatment (BMT) only.
Methods
SWITCH is an international multi-center randomized trial. 300 patients will be randomized (1:1) into either DC and BMT or BMT alone. Main inclusion criteria are spontaneous ICH of deep origin, NIHSS≥10 and ≤30, GCS > 7 and <14 and ICH volume > 30 and <100. The primary endpoint is severe disability and mortality, measured with the modified Rankin score 6 months after ictus.
Results
Recruitment started in October 2014. We currently have 33 sites open in Switzerland and in 7 other European countries. 97 patients were so far successfully randomized into the trial.
Conclusions
After initiation of SWITCH, we could increase the number of open centers to 33 and currently 97 patients have been randomized into the SWITCH trial. Further centers are still highly welcome and we believe that maintaining a high recruitment will be key for successful completion of the trial.
Trial registration number
NCT02258919
AS36-062
SOLITAIRE™ WITH THE INTENTION FOR THROMBECTOMY PLUS INTRAVENOUS T-PA VERSUS DIRECT SOLITAIRE™ STENT-RETRIEVER THROMBECTOMY IN ACUTE ANTERIOR CIRCULATION STROKE (SWIFT DIRECT)
1University Hospital of Bern, Department of Neurology, Bern, Switzerland
2University of Toronto, Department of Medical Imaging, Toronto, Canada
3Alfried Krupp Krankenhaus, Department of Neuroradiology, Essen, Germany
4University of Buffalo, Department of Neurosurgery, Buffalo, USA
5Vanderbilt University, Department of Neurology, Nashville, USA
6University of Toulouse, Department of Neuroradiology, Toulouse, France
7University Hospital Cleveland Medical Center, Department of Neurology, Cleveland, USA
8University of California Los Angeles, Department of Neurology, Santa Monica, USA
9University Hospital of Bern, Department of Neuroradiology, Bern, Switzerland
Background and Aims
Whether pre-treatment with intravenous thrombolysis (IVT) prior to mechanical thrombectomy (MT) with stent retrievers is beneficial has become a matter of debate. In a patient-level pooled analysis of five randomized controlled studies (HERMES collaboration) the treatment effect size of MT did not differ between patients receiving IVT and those treated with MT alone. SWIFT DIRECT aims to determine, whether direct MT in patients with proximal vessel occlusion in the anterior circulation is non-inferior to IVT and MT.
Methods
The international, multicentre, randomised-controlled, two-arm, open label, blinded endpoint (PROBE) trial SWIFT DIRECT will randomize 404 patients into the experimental arm (direct MT; 202) or control arm (bridging thrombolysis; 202). The trial will only be performed in patients with immediate access to MT. Main inclusion criteria are signed informed consent, age > 18 and <86 years, confirmed ischaemic stroke, NIHSS ≥8 and <30 and eligibility for IVT and MT. The primary outcome is functional independence (mRS) ≤2 at 90 days. Main secondary outcomes are mortality, change in NIHSS score post randomization, time to reperfusion (TICI ≥2b) and quality of life.
Results
The recruitment started end of 2017. Forty-eight patients have been enrolled by 25.03.2019. Over 30 sites in Switzerland, Germany, Austria, France, Spain, Finland and Canada will participate in this clinical trial.
Conclusions
If direct MT in patients with large artery vessel occlusion in the anterior circulation is as safe and efficacious as IVT and MT, this would have a relevant impact on future stroke management.
Trial registration number
ClinicalTrials.gov Identifier: NCT03192332
AS36-063
A REDUCTION IN TIME WITH ELECTRONIC MONITORING IN STROKE (ARTEMIS): A RANDOMIZED MULTICENTER TRIAL
1Amsterdam UMC- University of Amsterdam, Neurology, Amsterdam, The Netherlands
2Leiden University Medical Center, Neurology, Leiden, The Netherlands
3RAV Hollands Midden, Emergency Medical Services, Leiden, The Netherlands
4Isala, Neurology, Zwolle, The Netherlands
5Leiden University Medical Center, Clinical Epidemiology, Leiden, The Netherlands
6Julius Center- University Medical Center Utrecht, Clinical Epidemiology, Utrecht, The Netherlands
Background and Aims
Time is the most crucial factor for the clinical benefit of intravenous thrombolysis (IVT) and endovascular treatment (EVT). The delay from notification at the Emergency Medical Services (EMS) dispatch office until initiation of acute reperfusion therapy depends on logistics and collaboration between various caregivers in this trajectory (identified as ‘total system delay’ [TSD]). We aim to evaluate whether providing real-time audio-visual feedback to caregivers will reduce TSD.
Methods
The ‘A Reduction in Time with Electronic Monitoring In Stroke’ (ARTEMIS) trial is a multiregional, multicenter, randomized open end-point ongoing trial. We include consecutive, adult patients considered for IVT and/or EVT by the EMS dispatch office or paramedic on-site in three regions in the Netherlands with a total caption area of 2.625.000 inhabitants. Real-time audio-visual feedback on actual TSD is provided to caregivers through an automatic tracking system, using patient-specific Bluetooth wristbands and pre-mounted tablets throughout the entire trajectory. Randomization of real-time audio-visual feedback is per patient. We will include 75 EVT- and 225 IVT-patients in each arm (real-time audio-visual feedback vs. regular care), in order to demonstrate a 20-minute reduction on TSD to EVT and a 10-minute reduction on TSD to IVT.
Results
At present, we included 16 patients (12 IVT and 4 EVT).
Conclusions
The ARTEMIS trial will investigate whether providing real-time audio-visual feedback to caregivers within the acute stroke trajectory will reduce TSD to IVT and/or EVT.
Trial registration number
Clinical Trial Registration: www.clinicaltrials.gov (NCT02808806).
AS36-064
MICRORNA AS DIAGNOSTIC BIOMARKER TO DIFFERENTIATE ACUTE ISCHEMIC STROKE FROM INTRACEREBRAL HAEMORRHAGE AND STROKE MIMICS: AN OBSERVATIONAL MULTICENTER STUDY
1Leiden University Medical Center, Neurology, Leiden, The Netherlands
2Leiden University Medical Center, Vascular Surgery and Einthoven Laboratory for Experimental Vascular Medicine, Leiden, The Netherlands
3Haaglanden Medical Center, Neurology, The Hague, The Netherlands
4Leiden University Medical Center, Emergency Medicine, Leiden, The Netherlands
5Medical University of Vienna, Laboratory Medicine, Vienna, Austria
Background and Aims
The diagnosis of acute ischemic stroke (AIS) is time-consuming and challenging with consequent delays to- and overtreatment with reperfusion therapies. Circulating microRNAs (miRNAs) are promising biomarkers in AIS, but were not yet studied in patients suspected of acute stroke by emergency medical services (EMS) paramedics. We want to investigate whether expression profiles of extravesicular (EV) miRNAs in the circulation can distinguish AIS from intracerebral haemorrhage (ICH) and stroke mimics in a clinical setting.
Methods
This multicenter observational study enrolls 120 patients presented by EMS paramedics with suspected stroke <6 hours after symptom onset. Patients will be included in three groups (each N = 40) with the final diagnosis of AIS, ICH or a stroke mimic. Blood samples on admission, at 24 and 48 hours and after ≥6–8 weeks will be collected. The final diagnosis will be determined based on clinical and radiological assessments. Differentially expressed circulating EV miRNA profiles will be identified by RNA-Sequencing, followed by real-time quantitative polymerase chain reaction (rt/qPCR) analysis. By calculating the area under the receiver operating characteristic curve, the capability of circulating EV miRNAs to distinguish AIS patients from ICH or stroke mimics will be analyzed.
Results
The first patient was enrolled in November 2018. The current inclusion number is 45 patients.
Conclusions
Our study will investigate if the speed and accuracy of acute stroke diagnosis will be improved by identifying circulating EV microRNAs profiles that could be readily translated to a point-of-care test in the clinic.
Trial registration number
N/A
AS36-065
MULTICENTER RANDOMIZED CLINICAL TRIAL OF ENDOVASCULAR TREATMENT OF ACUTE ISCHEMIC STROKE IN THE NETHERLANDS FOR LATE ARRIVALS: MR CLEAN-LATE
1Maastricht University Medical Center, Neurology, Maastricht, The Netherlands
2Maastricht University Medical Center, Radiology, Maastricht, The Netherlands
3Leiden University Medical Center, Radiology, Leiden, The Netherlands
4Haaglanden Medical Center, Radiology, The Hague, The Netherlands
5University Medical Center Groningen, Neurology, Groningen, The Netherlands
6Sint Antonius Hospital, Neurology, Nieuwegein, The Netherlands
7Amsterdam University Medical Center, Radiology, Amsterdam, The Netherlands
Background and Aims
Level 1A evidence exists for the efficacy of endovascular therapy (EVT) of acute ischemic stroke due to a proximal occlusion of the anterior intracranial circulation, started within 6 hours from symptom onset. Recently, EVT has been proven effective beyond the 6 hour time window in a highly selected population based on perfusion imaging. We hypothesize that EVT is safe and effective for patients treated between 6 and 24 hours after symptom onset or last seen well after selection based on presence of collateral flow on CT angiography (CTA).
Methods
MR CLEAN-LATE is a multicenter, prospective, randomized, open-label, blinded-endpoint trial, aiming to determine the safety and efficacy of EVT between 6 and 24 hours after symptom onset in acute ischemic stroke due to a proximal intracranial anterior circulation occlusion (distal intracranial carotid artery, M1, M2). We plan to enroll 500 patients. Only patients with preserved collateral flow on CTA will be included. Patients with severe clinical deficit (NIHSS > 10) and small infarct core volumes (<25ml) based on perfusion imaging will be excluded as they are eligible for EVT as standard care. The primary endpoint is the modified Rankin Scale score (mRS) at 90 days. Treatment effect will be estimated with ordinal logistic regression (shift analysis) on the mRS score. Secondary outcomes include mortality at 90 days, stroke severity at 24 hours and 5-7 days, intracranial hemorrhage, recanalization at 24 hours and final infarct size at 5-7 days.
Results
Inclusion started in January 2018. Currently 96 patients were included in 11 centers.
Conclusions
.
Trial registration number
NL58246.078.17; ISRCTN19922220
AS36-066
MR CLEAN-NO IV: INTRAVENOUS TREATMENT FOLLOWED BY ENDOVASCULAR TREATMENT VERSUS DIRECT ENDOVASCULAR TREATMENT FOR ACUTE ISCHEMIC STROKE CAUSED BY A PROXIMAL INTRACRANIAL OCCLUSION
1Amsterdam UMC – location Academic Medical Center, Neurology, Amsterdam, The Netherlands
2Amsterdam UMC – location Academic Medical Center, Radiology and Nuclear Medicine, Amsterdam, The Netherlands
3Maastrucht University Medical Center, Neurology, Maastricht, The Netherlands
4Maastrucht University Medical Center, Radiology, Maastricht, The Netherlands
5Haaglanden Medical Center, Neurology, The Hague, The Netherlands
6Haaglanden Medical Center, Radiology, The Hague, The Netherlands
7Catharina Hospital, Neurology, Eindhoven, The Netherlands
8Catharina Hospital, Radiology, Eindhoven, The Netherlands
9Erasmus Medical Center, Radiology, Rotterdam, The Netherlands
10Erasmus Medical Center, Neurology, Rotterdam, The Netherlands
Background and Aims
Several trials have shown that endovascular treatment (EVT) following intravenous alteplase (IVT) improves outcome of patients with acute ischemic stroke and a proximal intracranial occlusion. A meta-analysis of five randomized trials (Hermes collaboration, Lancet 2016), which also included patients with contraindications for IVT, could not show an effect of IVT before EVT, though this concerned observational data. The question arises whether IVT is beneficial in patients eligible for EVT.
Methods
The MR CLEAN-NO IV trial is a multicenter, prospective, randomized, open-label, blinded-endpoint trial, comparing IVT followed by EVT with direct EVT in patients with a confirmed occlusion of the distal intracranial carotid artery, M1 or proximal M2. We aim to include 540 patients. The primary endpoint is the modified Rankin Scale score (mRs) at 90 days. Secondary endpoints include eTICI score, and Barthel score at 90 days. Safety endpoints include symptomatic intracranial hemorrhage and embolization in a new territory on angiography during EVT. We will perform subgroup analyses for occlusion location, collateral score and thrombus perviousness, among others.
Results
Currently 135 patients (25%) have been included in 12 recruiting centers.
Conclusions
The MR CLEAN NO-IV trial will provide robust data on whether IVT is beneficial to patients who are eligible for EVT and who present directly to a comprehensive stroke center.
Trial registration number
ISRCTN80619088
AS36-067
THE LACUNAR INTERVENTION TRIAL 2 (LACI-2). A PROSPECTIVE RANDOMISED OPEN LABEL BLINDED ENDPOINT PARTIAL FACTORIAL TRIAL TESTING CILOSTAZOL AMD/OR ISOSORBIDE MONONITRATE IN LACUNAR STROKE
1University of Edinburgh, Centre for CLinical Brain Sciences, Edinburgh, United Kingdom
Background and Aims
25% of ischaemic strokes are lacunar, due to intrinsic cerebral small vessel disease (SVD). There are no specific secondary prevention treatments. Licensed drugs, isosorbide mononitrate (ISMN) and cilostazol have relevant effects and data. LACI-1 found the drugs were tolerated individually and together without safety concerns in 57 patients.
LACI-2 aims to assess feasibility of recruitment, tolerability and safety of both drugs given for one year, confirm event rates, and estimate power for a phase III trial.
Methods
LACI-2 is a prospective, randomised, open-label, blinded-endpoint, multicentre, 2x2 partial factorial trial. 400 patients are being recruited in 25 UK centres. Interventions are oral ISMN 25mg bd, cilostazol 100mg bd, both, or neither. Patients with clinically evident lacunar ischaemic stroke and compatible brain MR or CT are eligible. LACI-2 is powered for safety to detect deaths of 2.0% p.a. and will stop if deaths reach 4%. The primary endpoint is feasibility of randomisation and follow-up of 400 patients across 25 sites over two years. Secondary outcomes are tolerability, safety (haemorrhage, death), recurrent stroke, dependency, cognitive impairment and SVD on MRI at one year. The British Heart Foundation funds LACI-2 with support from the Alzheimer’s Society UK.
Results
LACI-2 has recruited 120 patients to date; 25 centres are active. The Data Safety Monitoring Committee have encouraged recruitment.
Conclusions
The trial is on target to complete recruitment and one year follow-up of 400 patients by January 2021, preparatory to a large Phase 3 trial aiming to prevent recurrent lacunar stroke, physical and cognitive impairment.
Trial registration number
LACI-2 is registered ISRCTN14911850.
AS36-069
THE TRIPLE THERAPY PREVENTION OF RECURRENT INTRACEREBRAL DISEASE EVENTS TRIAL (TRIDENT): CHARACTERISTICS, SAFETY AND TOLERABILITY OF THE “TRIPLE PILL”
1The George Institute for Global Health, Academic Project Operations, Newtown, Australia
2University of Kelaniya, Pharmacology- Faculty of Medicine, Colombo, Sri Lanka
3National Hospital of Sri Lanka, Neurology, Colombo, Sri Lanka
4Linkou Chang Gung Memorial Hospital, Neurology, Taipei, Taiwan R.O.C
5University Kebangsaan Malaysia Medical Centre, Internal Medicines, Kuala Lumpur, Malaysia
6Radboud University Medical Centre, Neurology, Nijmegen, Netherlands Antilles
7University of Edinburgh, Clinical Brain Sciences, Edinburgh, United Kingdom
8University of Sydney, Medicine and Health, Westmead, Australia
9The George Institute for Global Health, Professorial Unit, Newtown, Australia
10The George Institute for Global Health, Neurology, Newtown, Australia
11The George Institute of China at Peking University, Health Science Centre, Beijing, China
Background and Aims
The incidence and prognosis of acute intracerebral haemorrhage (ICH) is linked to blood pressure (BP) levels. Intensive BP lowering is beneficial in preventing recurrent ICH. Many patients require more than one BP lowering agents to obtain effective BP control and adherence when taking multiple agents is poor. TRIDENT uses a fixed, low-dose combination in a single “Triple Pill” (telmisartan 20 mg, amlodipine 2.5 mg and indapamide 1.25 mg) on top of standard care in post ICH patients to determine the effectiveness of more intensive long term BP lowering on time to the first occurrence of recurrent stroke.
Methods
An international, multi-centre, double blind, placebo controlled parallel group, randomised, controlled trial involving 4200 patients from 100+ sites globally. Single-blind initial active run-in period, followed by double blind randomisation to active or placebo treatment with 6-monthly follow-up for an average of 3 years. Vital signs, BP, Na+, K+, serum creatinine, liver function and adverse events are collected at baseline, end of run-in, 6 weeks and at completion of study
Results
Recruitment commenced in September 2017, with current participation from hospitals across Australia, Netherlands, UK, Sri Lanka, Malaysia and Taiwan. Baseline characteristics, will be presented to show adherence, safety and tolerability of the “Triple Pill” in the first 200 participants enrolled.
Conclusions
Use of the “Triple Pill” in BP lowering post-ICH has good adherence, is safe and well tolerated across the participants.
Trial registration number
ClinicalTrials.gov – (NCT02699645)
AS36-070
PROTOCOL TO ASSESS THE COMBINED EFFECT OF ROBOTIC REHABILITATION AND TRANSCRANIAL DIRECT CURRENT STIMULATION ON PROPRIOCEPTION IN CHRONIC STROKE: A PILOT TRIAL
1University of Calgary, Clinical Neurosciences, Calgary, Canada
2University of Calgary, Hotchkiss Brain Institute, Calgary, Canada
3University of Calgary, Faculty of Kinesiology, Calgary, Canada
4University of Calgary, Department of Paediatrics, Calgary, Canada
Background and Aims
Proprioceptive deficits occur in ∼50% of individuals who have a stroke. Despite high prevalence rates, evidence-based interventions to improve proprioception are lacking and deficits often go untreated. Robotics and non-invasive brain stimulation are two exciting technologies that may help improve function after stroke. This single-blinded, randomized, sham-controlled pilot trial aims to determine whether robotic rehabilitation and transcranial direct current stimulation (tDCS) improves proprioception post-stroke.
Methods
Thirty individuals with chronic proprioceptive deficits are being randomized into three equally-sized groups: (1) robotic rehabilitation plus active tDCS, (2) robotic rehabilitation plus sham tDCS and (3) standard of care rehabilitation. Robotic rehabilitation consists of 10 extra hours of therapy across a 10-day period (1 hour per day). Therapy is conducted using the KINARM Exoskeleton and comprises of both active and passive training tasks that specifically target proprioception. The two treatment groups also receive either 20 minutes of 2mA anodal tDCS over the ipsilesional somatosensory cortex or sham tDCS. Stimulation is applied during the first 20 minutes of each therapy session. The primary outcome measure will be a validated measure of proprioception conducted by a robotic system (KINARM Exoskeleton). Secondary outcomes include various clinical assessment scores (Upper-Extremity Fugl-Meyer Assessment, Nottingham Sensory Assessment, Functional Independence Measure) and a robotic assessment of visually-guided reaching. Assessments are conducted pre- and post-intervention, and at a 3-month follow up. Assessments are performed by a blinded assessor therapist.
Results
Not yet available, trial ongoing.
Conclusions
This trial will explore potential rehabilitation techniques that may improve proprioception, thus enhancing functional outcomes post-stroke.
Trial registration number
ClinicalTrials.gov Identifier: NCT03888326
AS36-071
ONGOING TRIAL UPDATE 2019: DETERMINING OPTIMAL EARLY REHABILITATION AFTER STROKE (AVERT DOSE)
1Florey Institute of Neuroscience and Mental Health, Stroke Theme, Heidelberg, Australia
2University of Melbourne, Department of Medicine Austin Health, Heidelberg, Australia
3University of Glasgow, Institute of Cardiovascular and Medical Sciences, Glasgow, United Kingdom
4Christian Medical College, Department of Neurology, Ludhiana, India
5Monash University, Eastern Health Clinical School, Box Hill, Australia
6Monash University, Peninsula Clinical School, Clayton, Australia
7University of Newcastle, School of Health Sciences and Priority Research Centre for Stroke and Brain Injury, Newcastle, Australia
8Deakin University, Deakin Health Economics, Burwood, Australia
9Australian Catholic University, Nursing Research Institute, Sydney, Australia
10Monash University, Department of Medicine- School of Clinical Sciences at Monash Health, Clayton, Australia
11Auckland District Health Board, Allied Health, Auckland, New Zealand
12Norwegian University of Science and Technology, The Institute for Neuromedicine, Trondheim, Norway
13Singapore General Hospital, Physiotherapy Department, Singapore, Singapore
14University of South Australia, Sansom Institute for Health Research, Adelaide, Australia
15University Kebangsaan Malaysia, Department of Physiotherapy- Hospital Canselor Tuanku Muhriz, Kuala Lumur, Malaysia
16University of Sydney, Sydney Medical School, Sydney, Australia
17University of Melbourne, Translational Neuroscience, Parkville, Australia
18University of Melbourne, Melbourne Brain Centre, Parkville, Australia
Background and Aims
Our large international RCT of early rehabilitation (AVERT), provided evidence that early high intensity training interferes with stroke recovery (The Lancet, 2015), and that most patients may be responsive to therapy if the right dose is provided. (Neurology 2016). The aim of AVERT DOSE is to define the optimal early training regimens for people with mild and moderate ischaemic stroke.
Methods
Multi-Arm, Multi-Stage, Covariate-Adjusted, Response-Adaptive, randomised trial in two specified stroke severity strata. (Mild: NIHSS 0-7; Moderate: NIHSS 8-16). Patients are randomised to one of four mobility training regimens in each strata (including a pre-specified reference group), and the intervention is delivered for up to 14 days. Inclusion criteria: Ischaemic stroke within 48 hours, ≥18 years. Exclusion criteria: Severe stroke, medically unwell, no evident mobility problems. We will recruit 2,700 patients from 8 countries. Primary Outcome: Identification of the intervention regimen that results in fewer disabled patients (mRS 0-2) at 3 months post-stroke. Blinded assessments will occur at 3 and 6 months. An adaptive sample size re-estimation provides 80% power to detect a 10% absolute treatment effect or larger compared to the pre-specified reference group, with a significance threshold of p = 0.025 per stratum. Analyses will be intention-to-treat.
Results
Australian National Mutual Acceptance HREC approval for Australia has been received. International collaborations, site selection and development of an online database are underway.
Conclusions
AVERT DOSE will provide information about the dose and timing of early rehabilitation after ischaemic stroke onset. The results will be generalisable globally.
Trial registration number
UTN U1111-1221-2442
AS36-072
MULTICENTRE RANDOMISED TRIAL OF ACUTE STROKE TREATMENT IN THE AMBULANCE WITH A NITROGLYCERIN PATCH (MR ASAP)
1University Medical Center Utrecht, Neurology, Utrecht, The Netherlands
2Academic Medical Center Amsterdam, Neurology, Amsterdam, The Netherlands
3Isala, Neurology, Zwolle, The Netherlands
4Ambulance Ijsselland, Ambulance, Overijssel, The Netherlands
5Rijnstate, Neurology, Arnhem, The Netherlands
6Albert Schweitzer Ziekenhuis, Neurology, Dordrecht, The Netherlands
7Leiden University Medical Center, Neurology, Leiden, The Netherlands
8Radboud Medical Center, Neurology, Nijmegen, The Netherlands
9Ambulance Amsterdam, Ambulance, Amsterdam, The Netherlands
Background and Aims
The early administration of transdermal glyceryl trinitrate (GTN), or nitroglycerin, may increase the chance of a favourable outcome after stroke, possibly through an increase in intracranial collateral blood flow and reduction in blood pressure. Recent studies have reported conflicting results about the effect of GTN in the first hours after stroke. The aim of MR ASAP is to investigate the effect of transdermal GTN, started within 3 hours of stroke onset in the prehospital setting, on functional outcome at 90 days in patients with acute ischaemic stroke or intracerebral haemorrhage.
Methods
Phase III, multicentre, randomised, open-label clinical trial with blinded end point assessment (PROBE). 1400 patients with suspected stroke and a systolic blood pressure ≥ 140 mmHg will be randomly allocated to transdermal GTN (5 mg/day), administered via a transdermal patch by paramedics in the prehospital setting within 3 hours of stroke onset and continued for 24 hours, or to standard care. This trial is conducted in the Netherlands and uses a deferred consent procedure. The primary outcome is functional outcome, assessed with the modified Rankin Scale at 90 days and analysed with ordinal logistic regression.
Results
As of March 2019, 183 patients have been enrolled into the trial, but inclusion has temporarily been halted for DSMB review after publication of the RIGHT-2 trial in February 2019. This trial, comparable to MR ASAP in its design, showed no effect of early GTN administration on functional outcome.
Conclusions
Continuation of MR ASAP is expected in May 2019.
Trial registration number
ISRCTN99503308
AS36-074
TREATMENT WITH EXENATIDE IN ACUTE ISCHAEMIC STROKE (TEXAIS) TRIAL: PROSPECTIVE, RANDOMISED, OPEN LABEL, BLINDED END-POINT STUDY OF EXENATIDE VS. STANDARD CARE IN POST STROKE HYPERGLYCAEMIA
1The Florey, Ambulance Victoria/Monash University, Melbourne, Australia
2Westmead Hospital – Univ of Sydney- Sydney- Australia, Endocrinology, Sydney, Australia
3Monash Univ- Melbourne- Australia, Eastern Health Clinical School, Melbourne, Australia
4Florey Institute of Neuroscience and Mental Health- Melbourne- Australia, Statistics and Decision Analysis Academic Platform, Melbourne, Australia
5Australian Catholic Univ & St Vincent’s Health- Sydney- Australia, St Vincent’s Hospital, Sydney, Australia
6The Florey- Melbourne- Australia-, Stroke, Melbourne, Australia
7The University of Melbourne Director of Diabetes- Austin Health, Diabetes- Austin Health, Melbourne, Australia
8University of New South Wales, The Sydney Partnership for Health- Education- Research & Enterprise SPHERE, Sydney, Australia
9The University of Sydney, Westmead Clinical School Faculty of Medicine and Health, Sydney, Australia
10University of Melbourne, Melbourne Brain Centre- Royal Melbourne and Austin Hospitals, Melbourne, Australia
11Melbourne University, Melbourne Brain Centre- Royal Melbourne Hospital, Melbourne, Australia
12Claire Muller- Royal Brisbane and Women’s Hosp- Brisbane- Australia, Neurology, Brisbane, Australia
Background and Aims
Post-stroke hyperglycaemia (PSH) occurs in up to 50% of patients presenting with acute ischaemic stroke (AIS). It reduces the efficacy of thrombolysis, increases infarct size, and worsens clinical outcomes. Insulin-based therapies have generally not been beneficial in treating PSH as they are difficult to implement, may cause hypoglycaemia, possibly increase mortality and worse clinical outcomes. Exenatide may be a safer, simple, and more effective alternative to insulin in AIS
Methods
TEXAIS is a 3-year, Phase 2, multi-centre, prospective, randomised, open label, blinded end-point (PROBE) trial comparing Exenatide to Standard of Care. It aims to recruit 528 patients with a primary end point of major neurological improvement1 at 7 days defined as a ≥8-point improvement in NIHSS score, or NIHSS 0-1. Secondary outcomes of hyper- and hypoglycaemia at 5 days and NIHSS and mRS at 90 days will be measured. The treatment arm will receive Exenatide 5μg subcutaneously twice daily. The control arm will receive standard stroke unit care. Continuous glucose monitors will track the dynamic variability of glucose.
Results
Recruitment in TEXAIS is continuing with over 70 patients enrolled to date
Conclusions
TEXAIS aims to show that Exenatide is safe and effective in the treatment of PSH. It has been designed to be highly generalisable with an ability to enroll a large percentage of patients with AIS, regardless of admission blood glucose level, diabetes status, or stroke severity, with very low risk of hypoglycaemia.
Trial registration number
ACTRN12617000409370
AS36-075
START OR STOP ANTICOAGULANTS RANDOMISED TRIAL (SOSTART) AFTER INTRACRANIAL HAEMORRHAGE
1University of Edinburgh, Centre for Clinical Brain Sciences CCBS, Edinburgh, United Kingdom
Background and Aims
SoSTART aims to determine whether starting full treatment dose oral anticoagulation (OAC) results in a beneficial net reduction of all serious vascular events compared with not starting OAC in adults surviving spontaneous (non-traumatic) symptomatic intracranial haemorrhage (ICH) with persistent/paroxysmal atrial fibrillation/flutter (AF).
Methods
Eligibility criteria for the main study: Spontaneous symptomatic ICH, AF and a CHA2DS2-VASc score ≥2. Sub-study criteria: Brain magnetic resonance imaging to assess cerebral small vessel disease biomarkers before randomisation. Participants are randomised with 1:1 allocation of intervention: comparator. Intervention: start long-term (≥1 year) full treatment dose OAC (either a non-vitamin K antagonist direct oral anticoagulant [DOAC] or vitamin K antagonist if a DOAC cannot be used), chosen before randomisation. Comparator: do not start OAC (antiplatelet drug[s] or no antithrombotic drugs). Primary outcomes: all symptomatic major vascular events (i.e. major adverse cardiac or cerebrovascular events [MACCE]) including non-fatal stroke, non-fatal acute coronary syndrome, vascular death, sudden death, or death of unknown cause. Secondary outcomes: individual symptomatic vascular events; individual types of fatal events; dependence according to the modified Rankin Scale. Follow-up: at least one year after randomisation, using postal/telephone questionnaires to primary care practitioners and participants/carers, including review of any medical records and brain scans relating to outcomes.
Results
We have completed the pilot phase including 60 participants. On 26 March 2019, 56 sites had recruited 83 participants.
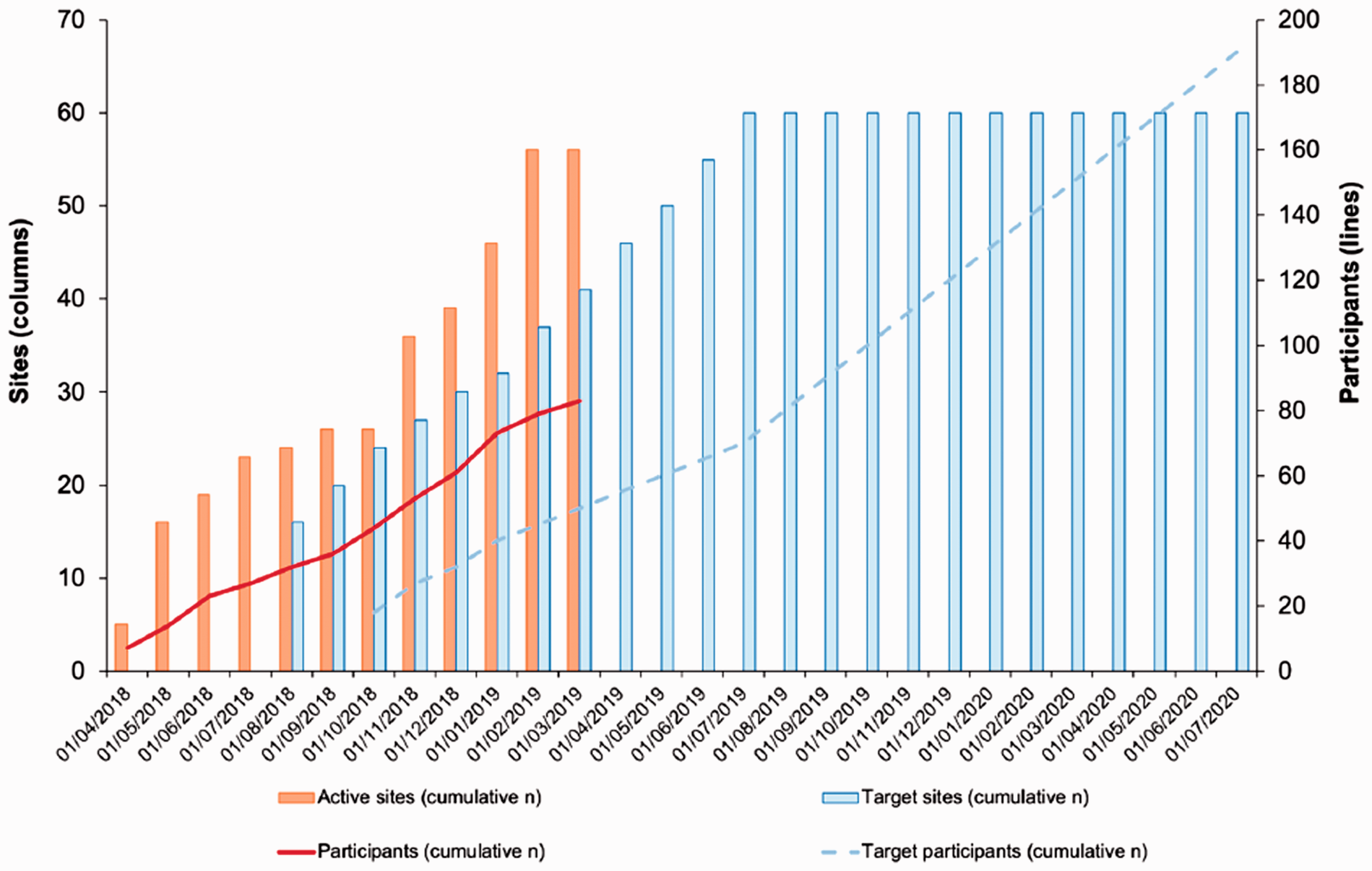
Conclusions
We will recruit at least 190 participants in the current safety phase by August 2020.
Trial registration number
Registration: NCT03153150; Website: www.SoSTART.ed.ac.uk
AS36-076
PREVENTION OF DEMENTIA USING MOBILE PHONE APPLICATIONS (PRODEMOS) – A MULTINATIONAL RANDOMIZED CONTROLLED TRIAL IN PROGRESS
1Amsterdam UMC/University of Amsterdam, Department of Neurology, Amsterdam, The Netherlands
2Amsterdam UMC/University of Amsterdam, Department of General Practice, Amsterdam, The Netherlands
3University of Toulouse UMR1027, Inserm, Toulouse, France
4Toulouse University Hospital, Department of Epidemiology and Public Health, Toulouse, France
5Brighton and Sussex Medical School University of Brighton, Department of Primary Care and Public Health, Brigthon, United Kingdom
6Karolinska Institutet Center for Alzheimer Research, Neurobiology- Care Science and Society, Stockholm, Sweden
7Cambridge Institute of Public Health/University of Cambridge, Department of Public Health and Primary Care, Cambrigde, United Kingdom
8Capital Medical University, School of Public Health, Beijing, China
9Alzheimer Europe, Executive Director, Luxembourg, Luxembourg
10VitalHealth Software, VitalHealth Software, Ede, The Netherlands
11Edith Cowan University, School of Medical and Health Sciences, Perth, Australia
12Radboud University Medical Centre, Department of Neurology – Donders Institute of Brain- Behaviour and Cognition, Nijmegen, The Netherlands
Background and Aims
The rising prevalence of dementia will largely occur in low- and middle-income countries. Up to 30% of all dementia is attributable to potentially modifiable risk factors. Mobile Health (mHealth) can improve accessibility to prevention and facilitate self-management.
Methods
Results
Conclusions
Trial registration number
N/A (pending)
AS36-077
TENECTEPLASE IN WAKE-UP ISCHAEMIC STROKE TRIAL (TWIST)
1Department of Clinical Medicine- UiT The Arctic University of Norway, Department of Neurology- University Hospital of North Norway, Tromsø, Norway
2Hospital of Southern Norway- Kristiansand, Department of Neurology, Kristiansand, Norway
3Oslo University Hospital, Department of Neurology, Oslo, Norway
4St Olavs Hospital, Department of Neurology, Trondheim, Norway
5Skåne University Hospital, Department of Neurology, Malmö, Sweden
6Uppsala University Hospital, Department of Neurology, Uppsala, Sweden
7Bispebjerg Hospital, Department of Neurology, Copenhagen, Denmark
8Rigshospitalet, Department of Neurology, Copenhagen, Denmark
9Tartu University Hospital, Department of Neurology and Neurosurgery, Tartu, Estonia
10Center for Neurology- Vilnius University, Department of Neurology and Neurosurgery, Vilnius, Lithuania
11Helsinki University Hospital, Department of Neurology, Helsinki, Finland
12University Hospital of Basel, Department of Neurology, Basel, Switzerland
13University of Leicester, Department of Cardiovascular Sciences, Leicester, United Kingdom
14UCL Institute of Neurology, Stroke Research Center, London, United Kingdom
15Oslo University Hospital, Department of Internal Medicine, Oslo, Norway
Background and Aims
Patients with wake-up stroke have traditionally been considered ineligible for intravenous thrombolytic treatment. Tenecteplase has pharmacological advantages over alteplase, and can be given as a bolus. We are performing a pragmatic, CT-based, randomised-controlled, open trial of tenecteplase for patients with wake-up stroke; the Tenecteplase in Wake-up Ischaemic Stroke Trial (TWIST).
Methods
Patients with wake-up stroke < 4.5 hours and without evidence of large infarct or intracerebral hemorrhage will be randomised to tenecteplase 0.25 mg/kg plus standard care or standard care alone. Plain brain CT and CT angiography will be done before randomisation and repeated on day 2. CT perfusion will be done at selected centres. Follow-up will be done at discharge (or day 7) and by telephone at 3 months.
The primary effect variable is functional outcome at 3 months, measured by the modified Rankin Scale.
Results
The target is to include 500 patients from centres in Norway, Sweden, Denmark, Finland, Estonia, Lithuania, UK, and Switzerland. Start of patient inclusion: June 2017. Status: 162 patients are included on the 26th of March. Study questions to be answered: 1. Can thrombolytic treatment with tenecteplase within 4.5 hours of wake-up improve functional outcome at 3 months? 2. Can findings on CT angiography or CT perfusion identify patients who benefit from such treatment, compared to patients without such findings?
Conclusions
TWIST will show whether patients with wake-up stroke can be treated with tenecteplase within 4.5 hours of awakening, and whether multi-modal CT can be used for identification of patients who benefit from treatment.
Trial registration number
ClinicalTrials.gov Identifier: NCT03181360
AS36-078
ALTEPLASE-TENECTEPLASE TRIAL EVALUATION FOR STROKE THROMBOLYSIS (ATTEST 2)
1University of Glasgow, Queen Elizabeth University Hospital, Glasgow, United Kingdom
2University of Glasgow, Robertson Centre for Biostatistics, Glasgow, United Kingdom
3University of Edinburgh, Centre for Clinical Brain Sciences, Edinburgh, United Kingdom
4Oxford University Hospitals NHS Trust, John Radcliffe Hospital, Oxford, United Kingdom
Background and Aims
Data from small randomised trials suggest that the modified tissue plasminogen activator tenecteplase at 0.25mg/kg is potentially superior to intravenous alteplase, the only medical treatment currently approved for acute ischaemic stroke. Significantly greater rates of early recanalization in large vessel occlusion were found in the EXTEND-IA TNK trial.
This pragmatic trial aims to test whether tenecteplase 0.25mg/kg is superior to alteplase using simple and universally available clinical and imaging criteria for patient selection. Even non-inferior safety and efficacy compared to alteplase would likely influence clinical practice as tenecteplase is significantly easier to administer than alteplase (and in many countries also less expensive).
Methods
ATTEST-2 will establish whether tenecteplase is superior to alteplase by undertaking a prospective randomized, open-label, blinded end-point (PROBE) trial in patients eligible for IV thrombolysis based on non-contrast CT imaging. Up to 60 UK centres will recruit 1870 patients.
Results
Status: All UK regulatory approvals are in place. The ATTEST-2 study has been adopted onto the NIHR Clinical Research Network Portfolio. Study recruitment is ongoing and will continue until Feb 2020.
Conclusions
An agent with superior risk:benefit ratio to alteplase would potentially extend thrombolytic treatment to a greater proportion of patients than at present and reduce the need for mechanical thrombectomy. This trial will contribute to the optimisation of reperfusion strategies.
Trial registration number
NCT02814409
AS36-079
CARDIOMYOCYTE INJURY FOLLOWING ACUTE ISCHEMIC STROKE (CORONA-IS) – RATIONALE AND DESIGN OF A PROSPECTIVE OBSERVATIONAL COHORT STUDY
1Charité Universitätsmedizin Berlin, Department of Neurology with Experimental Neurology, Berlin, Germany
Background and Aims
Myocardial injury (i.e. elevated cardiac troponin levels) is a frequent cardiac complication during the first few days after an ischemic stroke and is associated with a poor functional outcome. Myocardial injury represents one essential part of a broad spectrum of cardiac complications ranging to severe arrhythmia or heart failure. There is evidence that, in the majority of patients, the underlying mechanism of stroke-associated myocardial injury is not coronary-mediated myocardial ischemia but rather stroke-induced functional and structural interference in the central autonomic network. We hypothesize that this interference causes a dysregulation of normal neuronal cardiac control leading to myocardial edema and stunning (‘Stroke-Heart-Syndrome’).
Methods
CORONA-IS is a prospective, observational, single-centered cohort study that plans to recruit 300 patients with acute ischemic stroke. According to serial high sensitivity cTn levels during the first 24h after admission, patients will be assigned to three groups (no myocardial injury, chronic myocardial injury, acute myocardial injury). Study procedures include cardiovascular MRI and serial transthoracic echocardiography to visualize (transient) cardiac dysfunction and provide detailed tissue characterization, 20-minute Holter-monitoring with an analysis of specific autonomic markers, and a systematic biobanking to study further mechanisms such as altered microRNA signatures. A follow-up for cardiovascular events will be conducted one year after enrolment to study long-term effects of stoke-associated myocardial injury.
Results
N/A
Conclusions
The aim of the CORONA-IS study is to develop a better understanding of the characteristics and the pathophysiology of stroke-associated acute myocardial injury (‘Stroke-Heart-Syndrome’) in order to identify patients at risk and improve diagnostic and therapeutic procedures.
Trial registration number
EA4/123/18
AS36-080
IMPACT OF OMEGA-3 INDEX IN OUTCOMES OF STROKE: STUDY DESIGN
1University of Aberdeen, The Rowett Institute, Aberdeen, United Kingdom
2University of Aberdeen, Dept of Medicine & Therapeutics, Aberdeen, United Kingdom
Background and Aims
Stroke remains the main cause of adult long-term disability in the UK. Low serum n-3/n-6 polyunsaturated fatty acids (PUFAs) ratio and lower plasma levels of omega-3 PUFAs, particularly docosahexaenoic acid (DHA), predicts neurological deterioration in patients with acute ischaemic stroke and a higher ischaemic stroke severity on admission, respectively.
We aim to determine whether omega-3 Index, defined as the relative proportion of eicosapentaenoic acid (EPA) and DHA in erythrocytes, is associated with severity and functional outcome for both haemorrhagic and ischaemic strokes. Additionally, we will ascertain the feasibility of assessing dietary omega-3 PUFAs intake by recall in this population, and explore whether circulating fatty acid binding protein 4 (FABP4) could represent a useful predictor for stroke severity and recovery.
Methods
We are currently recruiting patients admitted to Aberdeen Royal Infirmary within 48 hours of stroke onset. Information on stroke type, subtype, and severity is recorded. Blood samples are obtained as close to ictus as feasible to determine Omega-3 Index, a second sample is collected in a fasting state to determine circulating FABP4 levels. Recovery is assessed using mRS at one and three months after stroke onset. Information on dietary intake of omega-3 PUFAs within three months prior to stroke is collected using a Food Frequency Questionnaire.
CGAC is funded by the Mexican government (CONACYT and I2T2).
Results
Enrolment is ongoing, last follow-up expected by September 2019.
Conclusions
This study will provide insights into dietary intake of omega-3 PUFAs, omega-3 Index, and outcomes of stroke in a UK population.
Trial registration number
researchregistry4006
AS36-081
RATES, RISKS AND ROUTES TO REDUCE VASCULAR DEMENTIA (R4VAD)
1University of Edinburgh, Centre for Clinical Brain Sciences, Edinburgh, United Kingdom
Background and Aims
Cognitive impairment and dementia are common complications of stroke, but limited knowledge about risk factors restricts mechanistic understanding, prevention, treatment and improvements in patient services. R4VAD is a multi-site longitudinal, inclusive study in patients presenting with stroke to UK stroke centres aiming to determine rates of, and risk factors for, cognitive and related impairments after stroke, assess mechanisms and improve prediction models.
Methods
R4VaD is recruiting patients within six weeks of stroke and collecting clinical, socioeconomic, lifestyle, cognitive, mood and informant data. More detailed assessments are obtained at 6+/-2 weeks post baseline assessment, with annual follow-up by phone and post to at least 2 years plus data linkage. Diagnostic neuroimaging is assessed in all, and high-sensitivity inflammatory blood markers and genetic analysis in as many patients as possible. Recruitment opened in September 2018 aiming to recruit 2000 patients by September 2020.
Results
To date, we have recruited 180 participants in 21 Centres in all four UK nations (mean age = 72.5 years, SD = 10.7; 54% female), 4-19 days post-stroke (mean = 9.0). So far, 85% of participants have ischaemic stroke; 6% ICH; 7% TIA; mean NIHSS = 2.8 (SD = 3.3); 10% lack capacity; 52% have an informant. Prevalent vascular risk factors include: hypertension (72%); hyperlipidaemia (47%); current/ex-smoker (59%); previous stroke/TIA (36%). At baseline, mean scores were: MOCA = 23.5/30, SD = 4.1;Zung depression = 47/80, SD = 13.6 (≥50 suggests depression) and anxiety (GAD-7) = 4.4/21, SD = 4.5 (≥5 suggests anxiety).
Conclusions
R4VAD will provide reliable data on cognitive and neuropsychiatric consequences long-term after stroke, improve understanding of clinical, demographic, laboratory, neuroimaging and social predictors of post-stroke cognitive impairment and dementia.
Trial registration number
n/a
AS36-082
PERFORMANCE FEEDBACK ON QUALITY OF CARE IN HOSPITALS PERFORMING THROMBECTOMY FOR ISCHEMIC STROKE (PERFEQTOS TRIAL)
1Erasmus University Medical Center, Neurology, Rotterdam, The Netherlands
2Erasmus University Medical Center, Public Health, Rotterdam, The Netherlands
3Amsterdam University Medical Center, Neurology, Amsterdam, The Netherlands
4Erasmus University Medical Center, Radiology and Nuclear Medicine, Rotterdam, The Netherlands
Background and Aims
Although provision of performance feedback on process indicators to health care professionals is common practice, observational studies of its effect on quality of care have shown mixed results. We propose an intervention study about the effect of performance feedback on quality of care for ischemic stroke.
Methods
PERFEQTOS is a stepped-wedge cluster randomized trial. All eighteen hospitals in The Netherlands providing endovascular treatment (EVT) for ischemic stroke will be included. Performance feedback consisting of 3-monthly reports with indicators on quality of care (case-mix, structure, process, outcome) in the own hospital for patients with ischemic stroke treated with EVT. This performance feedback is provided to local Quality Improvement Teams (QIT), including a neurologist, interventional neuroradiologist, neurology resident, and a stroke nurse. The QIT uses the performance feedback to define target(s) on (a) specific indicator(s) and to develop a Performance Improvement Plan (PIP). The impact of this PIP is evaluated in the next performance report. The control group will not receive structured performance feedback and is not required to have QIT. Primary outcome is the door-to-groin time (median), stratified for direct and transferred patients. Secondary outcomes include the door-to-needle time and 3-month mRS, adjusted for case-mix.
Results
Randomization of hospitals to cross from control to the intervention group will start in July 2019. Every six months 4 or 5 hospitals will be randomized and this will continue until all hospitals are crossed over.
Conclusions
We hypothesize that giving feedback to healthcare providers on the performance of their own hospital improves quality of stroke care.
Trial registration number
N/A
WITHDRAWN
AS36-086
APACHE-AF: APIXABAN VERSUS ANTIPLATELET DRUGS OR NO ANTITHROMBOTIC TREATMENT AFTER ANTICOAGULATION-ASSOCIATED INTRACEREBRAL HAEMORRHAGE IN PATIENTS WITH ATRIAL FIBRILLATION. A RANDOMISED PHASE II CLINICAL TRIAL
1Radboudumc
2UMC Utrecht
3UMC Groningen
4Albert Schweitzer Ziekenhuis
5Ziekenhuis Rijnstate
6Gelre Ziekehuizen
7Amphia Ziekenhuis
8Medisch Spectrum Twente
9Maastrich UMC
10Elisabeth Tweesteden Ziekenhuis
11Amsterdam MC
12Leiden UMC
13Zuyerland
14Onze Lieve Vrouwen Gasthuis
15Erasmus MC
16Haaglanden MC
Background
There is a lack of evidence on the optimal prevention of ischaemic stroke in patients with atrial fibrillation and a recent intracerebral haemorrhage (ICH) during treatment with oral anticoagulation. Treatment with a direct oral anticoagulant like apixaban might be an attractive alternative in terms of a lower risk of recurrent ICH than with a vitamin-K antagonist, while at the same time being effective for the prevention of ischaemic stroke.
Objective
To obtain reliable estimates of the rates of vascular death or non-fatal stroke in patients with atrial fibrillation and a recent anticoagulation-associated ICH who are treated with apixaban versus those who are not treated with oral anticoagulation.
Study design
Multi-centre, phase II, randomised, open-label clinical trial with blinded outcome assessment.
Study population
100 adults with a history of atrial fibrillation and ICH during treatment with oral anticoagulation in whom clinical equipoise exists on the optimal stroke prevention therapy.
Intervention
Patients are randomised to apixaban 5 mg twice daily or to avoiding anticoagulation. Patients will be randomized between 7 and 90 days after the index haemorrhage.
Primary outcome: Vascular death or non-fatal stroke during follow-up.
Sample size: Ten primary outcome events in 100 patient-years of follow up will yield a 95% confidence interval of 4.9 to 17.6.
Trial registration number
Registration: NTR4526; NCT02565693.