
Abstract
Introduction:
Common oral diseases are known to be associated with dysbiotic shifts in the supragingival microbiome, yet most oral microbiome associations with clinical end points emanate from cross-sectional studies. Orthodontic treatment is an elective procedure that can be exploited to prospectively examine clinically relevant longitudinal changes in the composition and function of the supragingival microbiome.
Methods:
A longitudinal cohort study was conducted among 24 adolescent orthodontic patients who underwent saliva and plaque sampling and clinical examinations at time points: before fixed appliance bonding and at 1, 6, and 12 wk thereafter. Clinical indices included bleeding on probing (BOP), mean gingival index (GI), probing depths (PDs), and plaque index (PI). To study the biologically (i.e., transcriptionally) active microbial communities, RNA was extracted from plaque and saliva for RNA sequencing and microbiome bioinformatics analysis. Longitudinal changes in microbiome beta diversity were examined using PERMANOVA tests, and the relative abundance of microbial taxa was measured using Kruskal–Wallis tests, Wilcoxon rank-sum tests, and negative binomial and zero-inflated mixed models.
Results:
Clinical measures of oral health deteriorated over time—the proportion of sites with GI and PI ≥1 increased by over 70% between prebonding and 12 wk postbonding while the proportion of sites with PD ≥4 mm increased 2.5-fold.
Conclusions:
This study offers insights into longitudinal community and species-specific changes in the supragingival microbiome transcriptome during fixed orthodontic treatment, advancing our understanding of microbial dysbioses and identifying targets of future health-promoting clinical investigations.
Knowledge Transfer Statement:
Bonding braces was associated with subsequent changes in the oral microbiome characterized by increases in disease-associated species, decreases in health-associated species, and worsened clinical measures of oral health.
Introduction
Dental plaque biofilms are composed of diverse microbial communities that play key roles in modulating oral health and disease. Although most oral diseases are now best understood as dysbiotic shifts (i.e., imbalances) in the oral microbiome, little is known about specific microbial-ecological events that precede the development of disease or support health. Moreover, most reported oral microbiome associations with oral or dental outcomes emanate from cross-sectional studies, limiting our ability to make causal inferences about the pathogenic function or predictive utility of identified species. Investigations of the taxonomic and biological mechanisms underpinning the natural history of oral and dental diseases are warranted to reveal yet-to-be identified oral pathogens and their polymicrobial interactions, as well as taxa that help maintain health-associated homeostasis. Prospective clinical investigations have the potential to decisively advance the oral microbiome knowledge base.
A well-recognized extrinsic factor that challenges oral health and can be leveraged in prospective clinical investigations involving the oral microbiome is the introduction of orthodontic fixed appliances. Fixed appliances present a challenge to maintaining oral hygiene, leading to an increase in plaque accumulation and elevated risk of caries, gingivitis, and periodontitis (Lu et al. 2018). Gingival inflammation, presenting as increased gingival bleeding and hyperplasia, is a common complication of orthodontic treatment (Pinto et al. 2017). Increases in periodontal pathogens within dental plaque biofilm drive this inflammation. Therefore, periodontal health is a logical clinical outcome to monitor and correlate with concomitant microbiome changes during orthodontics.
Technological advances now enable high-resolution studies of microbial composition and activity via next-generation sequencing (NGS) involving high-throughput DNA sequencing (DNA-seq) and RNA sequencing (RNA-seq). Previous reports of microbiome changes following orthodontic fixed appliances generally include 1) increase of key species’ abundance and decrease in the diversity of the oral microbiome following bonding and 2) a reestablishment of host–microorganism balance after 3 mo of treatment (Lucchese et al. 2018; Wang et al. 2019). While much has been learned, significant questions remain, as many earlier studies lacked comprehensive periodontal examinations. Others were based on saliva samples (rather than plaque) and investigated a select number of pathogenic species rather than the entire microbiome. We posit that there is a need for total RNA sequencing–based approaches to provide insight into species that are transcriptionally active (and arguably most clinically relevant), allowing the creation of a microbial community’s functional profile. Indeed, the use of RNA-seq to investigate microbial samples has enabled a richer understanding of species composition and activity in the oral cavity compared to culture-dependent study designs or the use of 16S ribosomal RNA (rRNA) gene sequencing technology (Zhang et al. 2021).
The present study sought to address these needs by concurrently evaluating microbiome shifts reflected in the RNA-seq transcriptome and clinical changes that occur following bonding of fixed appliances. To this end, orthodontic patients were enrolled in a prospective cohort study where a comprehensive periodontal evaluation was carried out, and saliva and supragingival plaque samples were collected immediately before bonding and at 1, 6, and 12 wk postbonding. RNA sequencing was performed on plaque samples to determine changes in the microbial transcriptomes and species diversity. RNA sequencing was also performed on baseline saliva samples to determine if relative species abundance at treatment start could be predictive of later clinical outcomes. We hypothesized that introduction of fixed appliances would lead to dysbiotic shifts of the supragingival biofilm and that would manifest as clinical indicators of periodontal inflammation as well as a commensurate increase in periodontal disease– and caries-associated pathogens.
Materials and Methods
Sample Recruitment
This study was approved by the University of North Carolina (UNC) Office of Human Research Ethics (IRB #16-1624) and conforms to Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines for cohort studies. Screened and consented individuals were recruited from the Orthodontics Clinic at the UNC School of Dentistry from 2017 to 2021 (Appendix Table 1). Data were collected by calibrated examiners at 4 time points: just prior to bonding of fixed appliances (T0) and then at 1 (T1), 6 (T2), and 12 (T3) wk after bonding (Fig. 1). Twenty-four healthy adolescents (ASA 1–2), of both genders, with no active caries or periodontitis as confirmed by their general dentist and orthodontist, receiving comprehensive, fixed orthodontic treatment were included if they completed at least 2 study visits (Table 1). When the project began, there was a lack of published data on effect size for microbiome differences to determine sample size; as a result, a convenience sample of 24 participants was selected and is comparable to those used in similar studies (Chen et al. 2022; Shokeen et al. 2022). Because of variability in appointment intervals and missed appointments, related in part to the COVID-19 shutdown, data were collected from subjects at T0 and at least 1 of the remaining time points.

Study summary. (
Participants’ Demographics.
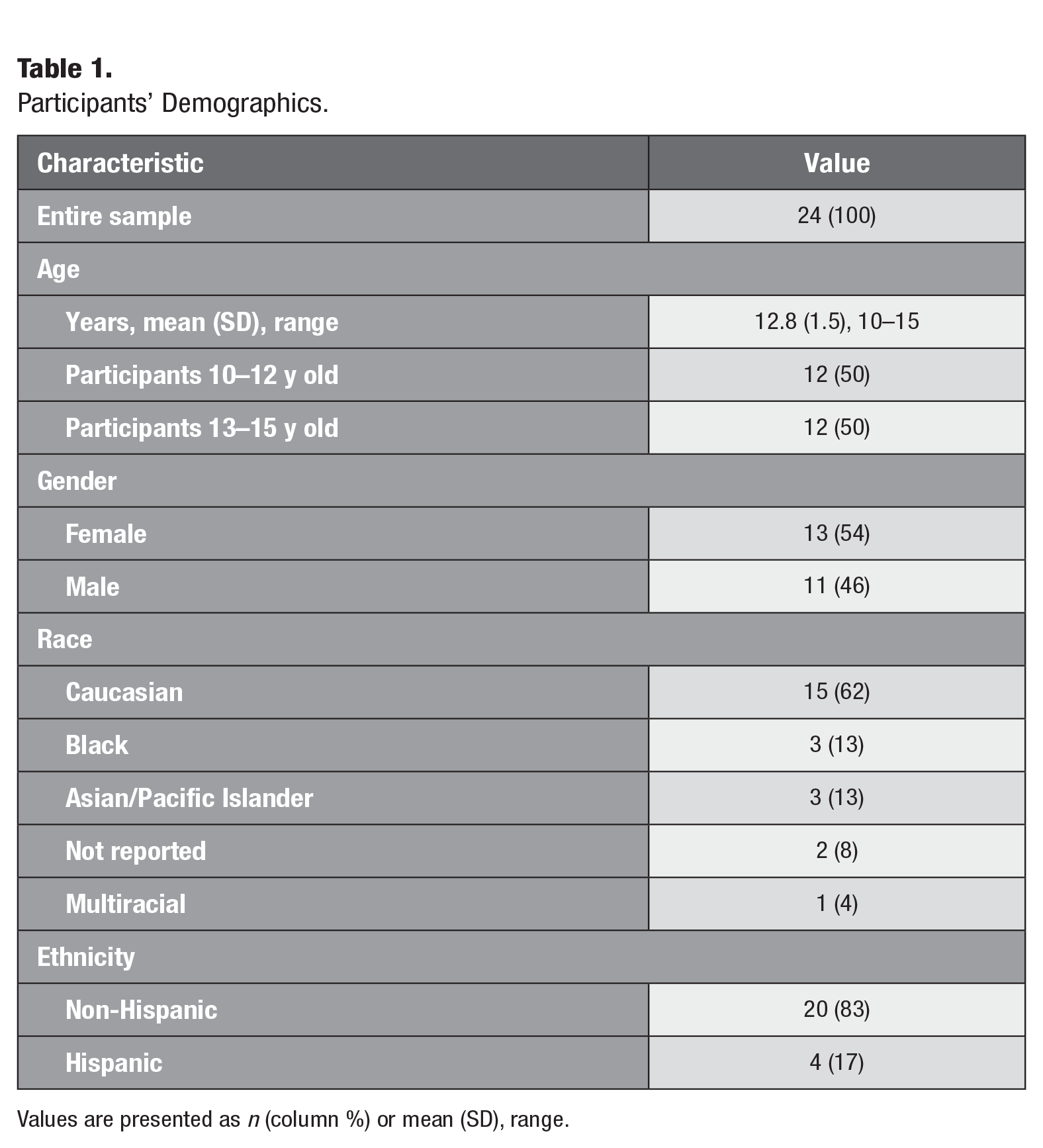
Values are presented as n (column %) or mean (SD), range.
Clinical Data Collection and RNA Extraction and Sequencing
At each visit, an unstimulated saliva sample and supragingival plaque samples were collected. Comprehensive periodontal evaluations were performed. Detailed protocols for data collection and RNA extraction and sequencing are included in the Appendix.
Bioinformatics Processing
Sequencing output was converted to fastq format and demultiplexed using Illumina Bcl2Fastq 2.20.0. (Illumina). Quality control (QC) was carried out using FastQC 0.11.8 (Babraham Institute). Adapters were trimmed using Trim Galore! 0.6.5 (Babraham Institute). The resulting paired-end reads were classified with Kraken2 (Wood et al. 2019) and Bracken 2.5 (Lu et al. 2017), and all reads identified as host were eliminated. Paired-end reads were joined with vsearch 1.10.2 (Rognes et al. 2016). The resulting single-end reads were again trimmed of remaining adapters using Trim Galore. Estimates of taxonomic composition and gene family, regrouped to Enzyme Commission (EC) number, were produced from the remaining reads using HUMAnN3 (Abubucker et al. 2012; Beghini et al. 2021). Raw data were deposited in the National Institutes of Health (NIH) Sequence Read Archive system as a BioProject submission entitled, “Longitudinal Microbiome Changes in Supragingival Biofilm Transcriptomes Induced by Orthodontics” (PRJNA994526).
Alpha diversity with respect to Shannon entropy with a Kruskal–Wallis test and beta diversity measured by Bray–Curtis dissimilarity with PERMANOVA indices were estimated using QIIME 2 (Bray and Curtis 1957) at a rarefaction depth of 5,000 sequences per subsample for estimates of taxonomy, gene families, and pathways longitudinally (Appendix Tables 2–3) (Caporaso et al. 2010). Results were summarized and visualized through principal coordinate analysis as implemented in QIIME 2.
A linear mixed model was used to determine differences in clinical measures, with repeated measures by visit (Fig. 2, Appendix Table 4) (Cnaan et al. 1997). Pairwise P values were computed using least square means from the linear mixed model, with Tukey adjustment for multiple testing. Clinical data were further stratified and compared longitudinally across strata of age (10–12 versus 13–15 y), gender (male versus female), and race (Caucasian versus non-Caucasian; data not shown). To study taxon-specific longitudinal changes in an unbiased manner, all available taxa were first filtered according to their detection frequency. By considering species with >10,000 assigned reads, 551 nonhost species were initially retained, and 522 of those occurred in ≥20% of samples. This set of 522 taxa was carried forward to downstream association analyses. To facilitate comparison with previous studies, results for 32 selected species from this list are reported based on their known roles in oral health and disease, including caries and periodontal disease (Zhu et al. 2018; Cho et al. 2023) (Appendix Table 5). Species abundance data were log-transformed and longitudinal differential abundance analysis was performed using negative binomial and zero-inflated mixed models (NBZIMM) (Zhang and Yi 2020). Three models were considered: including only visit as a fixed effect, including only bleeding on probing (BOP) as a fixed effect, and including both visit and BOP as fixed effects. For all 3 models, participants were modeled as a random effect. Missing values of BOP were replaced by the median value of all samples with observed BOP.
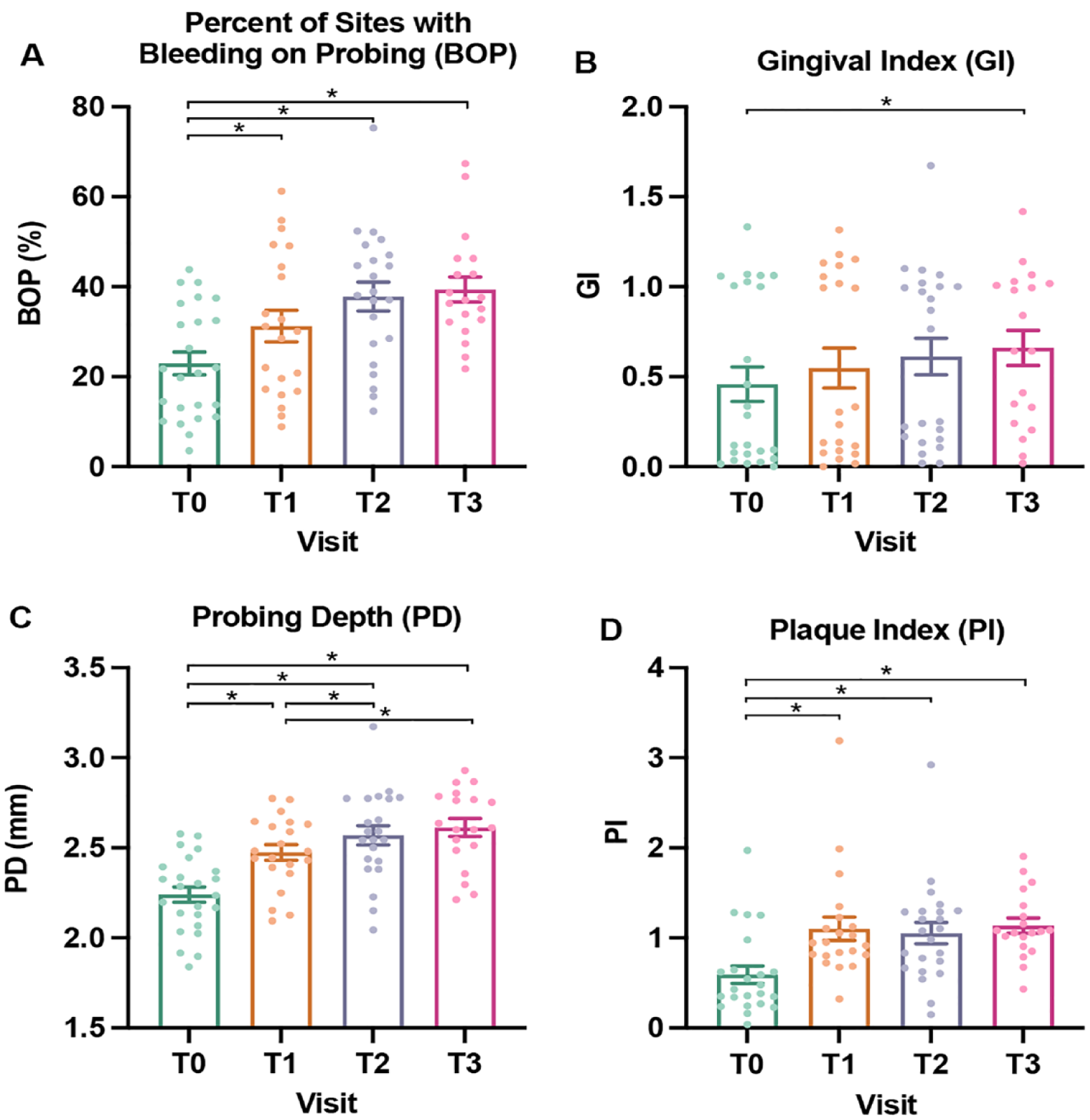
Longitudinal changes in measures of oral health. (
To test whether any microbial taxa measured at T0 were predictive of gingival inflammation at 12 wk postbonding, the cohort was divided into 2 groups: participants with BOP at ≥40% of sites (n = 7) and those who had BOP at <40% of sites (n = 14) at T3, 3 mo into treatment. The cutoff of ≥40% of sites with BOP was chosen as the criterion for “widespread gingival inflammation” compared to the cutoff of >30% of sites for “gingivitis.” Differences in relative species abundance between high (≥40% of sites) and low BOP (<40% of sites) groups were compared in both plaque and saliva samples.
Among species present at T0, differential abundance analysis using ALDEx2 and ANCOM-BC was performed to identify species associated with extensive gingival inflammation (i.e., ≥40% of sites with BOP) at T3. For ANCOM-BC, only species that had greater than 1,000 reads and occurred in at least 50% of the samples were considered. For species with an estimated ANCOM-BC q value less than 0.2, ALDEx2 was applied for a second estimate of differential abundance (Nearing et al. 2022). Finally, for each of the 32 literature-based prioritized species, abundance data were log-transformed and subjected to the Kruskal–Wallis tests. Significant differences between visits were determined by the Wilcoxon rank-sum test. The R package “ggplot2” was used to generate relevant visualizations.
Results
Twenty-four adolescents, ages 10 to 15 y (mean, 12.8 y), were enrolled and followed longitudinally, with equal gender distribution (Table 1). A third of participants had gingivitis (>30% of sites with BOP) at baseline, and 79% developed gingivitis at T3. Overall, measures of oral health worsened during the study period (Fig. 2, Appendix Table 4). Plaque depth (PD) and the proportion of sites with BOP steadily increased over time. These trends paralleled increases in mean plaque index (PI) and gingival index (GI). Meanwhile, immaterial changes (i.e., <0.1 mm) were found in Clinical Attachment Loss (CAL) and no differences in measures of oral health according to participants’ age, gender, and race (data not shown). Due to a lack of significant differences in these covariates, age, race, and gender are not included in the models. Additionally, the study is based on longitudinal data from the same patients across multiple time points, with a focus on comparisons between time points, such that age will not confound associations between microbiome changes and time.
RNA-seq of the plaque biofilm microbiome resulted in 725 million high-quality reads. A total of 414 genera were identified, of which 240 were assigned more than 1,000 reads, and 975 species were identified, of which 666 were assigned more than 1,000 reads. Although the mean percentage of taxa unclassified reads per sample was 11.4%, the mean per sample percentage of reads unmapped to a gene family by HUMAnN3 remained 44.2%. The range of microbial raw read pairs was 6,510 to 33,629,288, and the average was 8,442,916 (Appendix Table 6).
Five genera constituted 63% of reads at T0: Actinomyces, Streptococcus, Capnocytophaga, Arachnia, and Corynebacterium, within which 5 species constituted 27% of reads: Streptococcus pneumoniae, Actinomyces dentalis, Actinomyces massiliensis, Arachnia propionica, and Actinomyces sp. oral taxon 175 (Fig. 3A, B). Functionally, in the samples derived from plaque, 447 enzymes were assigned more than 5,000 reads. Metabolic enzymes, several linked to oral disease, were highly expressed, including NADPH/quinone reductase, peroxiredoxin, L-lactase dehydrogenase, pyruvate kinase, and superoxide dismutase, along with synthetases such as glutamine synthetase and DNA-directed RNA polymerases, needed for growth and homeostasis of bacteria (Fig. 3C, Appendix Table 7).
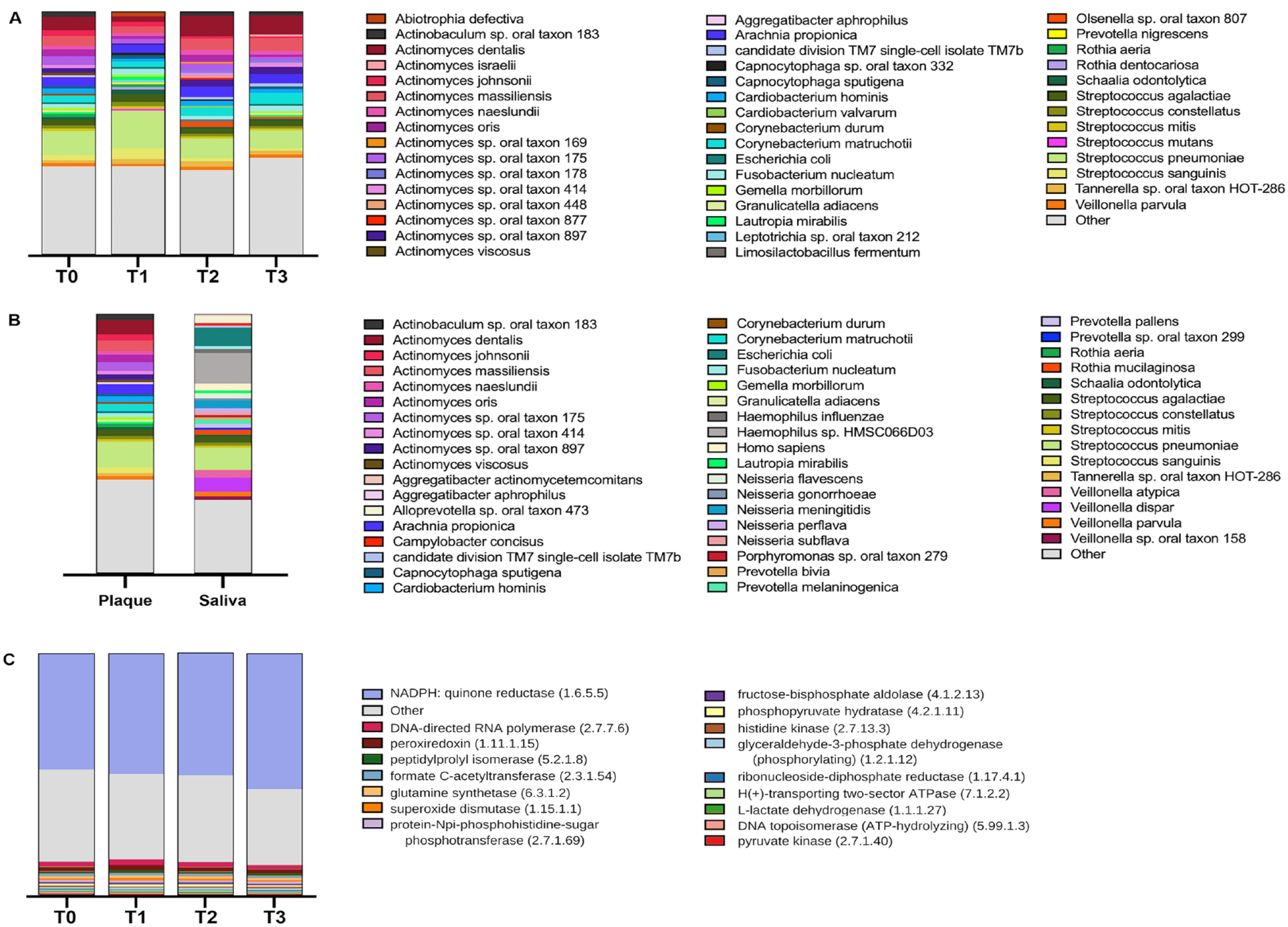
Diversity analyses of plaque and saliva. (
There were no longitudinal differences in the microbiome community structure for alpha diversity (P = 0.565, Appendix Fig. 1A, Appendix Table 2); however, differences were found for beta diversity over time (P = 0.016, Appendix Fig. 1B). Plaque beta diversity showed an initial nonsignificant (q = 0.208) decrease following bonding (T0 to T1) and then increased significantly (q = 0.018) at 6 wk (T1 to T2) and 12 wk thereafter (q = 0.018 T1 to T3) (Appendix Table 3).
Among the longitudinally analyzed species with >1,000 assigned reads, 19 species were found to vary significantly (adjusted P < 0.05) in relative abundance in plaque with respect to visit only and 4 were inversely associated with BOP (Appendix Table 8). Mogibacterium diversum and Mogibacterium timidum were associated with both longitudinal changes between visits and BOP (Fig. 4C). Four additional known periodontal and cariogenic pathogens or markers of dental health were identified among species showing significant change in relative abundance longitudinally over the visits (Fig. 4B, Appendix Fig. 3). Prevotella salivae and Selenomonas sputigena, identified as of interest a priori (Fig. 4A), but also, Olsenella sp. oral taxon 807 and Prevotella melaninogenica, exhibited an increase in abundance from T0 (prebonding) to T3 (12 wk postbonding). However, Actinomyces johnsonii and Streptococcus sanguinis showed a significant decrease in abundance from T0 to T3. Analogously, the association of relative abundance with both visit and BOP for the species of the genus Mogibacterium is shown in Figure 4C. All 3 species show a marked increase in relative abundance over time, although the rate of increase appears to be dependent on the amount of BOP. The overall increase in BOP with respect to visit is also of note. Conspicuously absent from the identified species with respect to visit or BOP is the well-known cariogenic species Streptococcus mutans. S. mutans did not vary in relative abundance over time.
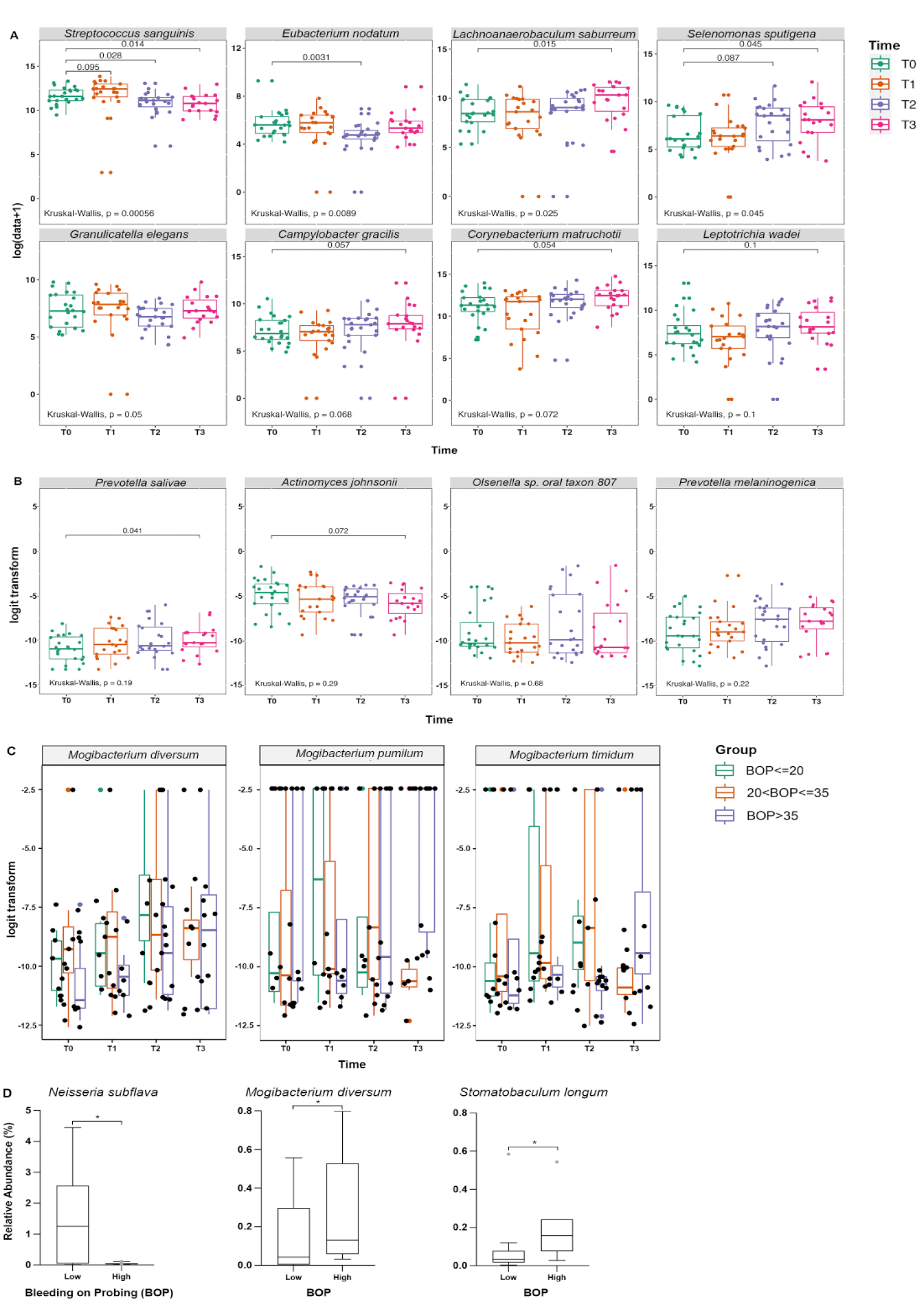
Bacterial species with significant differences. (
Among the 32 literature-identified periodontal and cariogenic pathogens that were selected (Appendix Table 5), 8 species showed significant changes in plaque over time (Fig. 4A, Appendix Fig. 2). Lachnoanaerobaculum saburreum, Selenomonas sputigena, Campylobacter gracilis, Corynebacterium matruchotii, and Leptotrichia wadei exhibited a steady increase in abundance from T0 to T3. In contrast, Eubacterium nodatum and Granulicatella elegans showed temporary decreases at T2 (Fig. 4A). Streptococcus sanguinis was the only species to show a significant, lasting decrease in abundance from T0 to T3.
Longitudinal analysis of predicted enzymes encoded by the biofilm and salivary transcriptomes revealed 245 enzymes exhibiting a significant variation with respect to visit (Appendix Table 7), but no observed longitudinal variations with respect to BOP were significant after multiple testing correction. Of the 245 enzymes identified as varying significantly in relative abundance with respect to visit, 244 exhibited a statistically significant reduction in relative abundance over time, the most significant being CCA tRNA nucleotidyltransferase (EC 2.7.7.72), phosphoglycerate mutase (EC 5.4.2.1), adenine phosphoribosyltransferase (EC 2.4.2.7), nonspecific serine/threonine protein kinase (EC 2.7.11.1), phosphatidate cytidylyltransferase (EC 2.7.7.41), ABC-type xenobiotic transporter (EC 3.6.3.44), phosphopantothenoylcysteine decarboxylase (EC 4.1.1.36), phosphopantothenate-cysteine ligase (CTP) (EC 6.3.2.5), diacylglycerol kinase (ATP) (EC 2.7.1.107), DNA topoisomerase (ATP-hydrolyzing) (EC 5.6.2.2), and dihydrolipoyl dehydrogenase (EC 1.8.1.4). Only 3-hydroxybutyryl-CoA dehydrogenase (EC 1.1.1.157) statistically significantly increased in relative abundance over time.
Differences in relative species abundance between high (≥40% of sites) and low (<40% of sites) BOP groups were examined in plaque and saliva microbiomes. No species found in plaque biofilm were predictive of later BOP measures. The relative abundance of Stomatobaculum longum and Mogibacterium diversum in prebonding (T0) saliva samples was found to be predictive of higher BOP 12 wk postbonding (T3), whereas Neisseria subflava was associated with lower BOP (Fig. 4D, Appendix Table 9).
Discussion
In this study, the natural history of biofilm changes during early orthodontic treatment was examined and clinical end points were correlated with community- and species-level microbiome RNA-seq data. Orthodontic treatment has a transitory negative impact on oral health during fixed appliance care and represents a unique clinical and real-life model for investigating early stages of periodontal disease and caries in an initially healthy patient cohort. The presence of brackets correlates with a worsening of nearly all clinical measurements of gingival health, concurrent with longitudinal microbial changes. The identified microbiome changes parallel what is expected in inflammatory and caries-associated dysbiotic shifts (Fig. 4).
In the present study, no significant changes in overall community alpha diversity were observed, despite changes in the relative abundance of specific microbial species (Appendix Fig. 1A, Fig. 4). This finding agrees with literature showing no difference in alpha diversity when comparing different orthodontic treatment modalities, irrespective of time point or sample location (Shokeen et al. 2022). A transient difference in community composition (beta diversity) was observed when comparing T1 to T2 and T3 (Appendix Fig. 1B). This trend in diversity at T1 could be due to the cleansing effect of acid etch at bonding of the orthodontic appliances 1 wk prior, causing compositional differences between T1 supragingival plaque microbiome and later time points (T2 and T3). The bacterial community highly expressed several metabolic enzymes linked to oral disease (Fig. 3C). Elevated L-lactase dehydrogenase activity in saliva and superoxide dismutase in gingival crevicular fluid have been associated with periodontal disease (De La Peña et al. 2007; Karim et al. 2012), while in children, pyruvate kinase activity is increased in dental plaque and correlates with caries progression (Krzyściak et al. 2017). Expression of NADPH/quinone reductase is important for bacterial colonization through oxidative stress resistance and could play a role in colonization in the oral cavity (Wang and Maier 2004). Similarly, S-glutathionylation, and in particular DNA topoisomerase I with 8 S-glutathionylated sites, although not necessarily DNA topoisomerase II, has been implicated in the cariogenicity of S. mutans (Li et al. 2020). Moreover, dihydrolipoyl dehydrogenase has been shown to be upregulated in coaggregations of S. gordonii and F. nucleatum, resulting in increased intracellular survival within macrophages of both species and attenuated production of proinflammatory cytokines IL-6 and IL-1β (Liu et al. 2022).
Longitudinal differences in differential species abundance were readily identified (Fig. 4). An important finding was the lasting decrease in the relative abundance of S. sanguinis from T0 to T3. S. sanguinis is a well-known marker of oral health, capable of antagonizing cariogenic pathogens such as S. mutans (Kreth et al. 2008). Absence of S. sanguinis has been associated with aggressive periodontitis (Stingu et al. 2008). Recent literature suggests S. mutans is associated with caries progression rather than causation; in our study, where one might expect new caries development (e.g., white spot lesions), S. mutans did not vary in relative abundance over time, consistent with it not being involved in caries initiation (Philip et al. 2018). Our data suggest a decrease in homeostatic, healthy bacteria like S. sanguinis and a rise in pathologic organisms, concurrent with worsening gingival health, suggesting species-level changes contribute to the pathogenesis of gingival inflammation and caries in orthodontic patients; such a cascade of microbiome changes may occur early in other oral disease states.
In contrast, E. nodatum and G. elegans showed a temporary decrease at T2. E. nodatum is a member of the “orange complex” and is closely related to progression of periodontal disease, evidenced by a significant correlation to increased PD and GI (Moon et al. 2018). G. elegans has been associated with early childhood caries (ECC) and white spot lesions during orthodontic treatment (Tanner et al. 2012). The cause of a temporary decrease in E. nodatum and G. elegans at T2 is unclear, but their observed rebound in abundance from T2 to T3 was expected with worsening oral health seen in orthodontic patients.
The relative abundance of L. saburreum, S. sputigena, C. gracilis, C. matruchotii, and L. wadei increased from T0 to T3. L. saburreum, S. sputigena, and L. wadei were identified as strongly associated with ECC development (Cho et al. 2023). S. sputigena and L. wadei have also been shown to dominate disease sites in generalized aggressive periodontitis (Faveri et al. 2008; Nibali et al. 2020). The pathogenicity of C. gracilis remains debated, as it is present in healthy sites at similar levels as in subjects with gingivitis and periodontitis (Liu et al. 2018). C. matruchotii is one of the most predominant bacteria found in dental plaque; it is considered a marker of a caries-free state yet appears to participate in deposition of supragingival calculus (Li et al. 2022).
P. salivae, Olsenella sp. oral taxon 807, P. melaninogenica, and S. sputigena exhibited increases in abundance across visits that correlated with clinical increases in BOP (Fig. 4A, B).P. melaninogenica is an anaerobic early colonizer of the infant mouth and ubiquitous in the oral cavity; P. salivae is also a common commensal oral bacterium, often found with P. melaninogenica (Könönen et al. 2022). P. salivae and P. melaninogenica have both been found in periodontitis, periodontal abscesses, ECC, and endodontic infections (Könönen et al. 2022). Increased Olsenella sp. oral taxon 807 has been associated with changes in the oral microbiome that occurred with the Neolithic transition from hunter-gathering to farming, with unclear implications for our patient base (Quagliariello et al. 2022). Finally, S. sputigena was recently identified as a new pathobiont for dental caries (Cho et al. 2023).
Our findings parallel those seen in studies investigating oral microbiota changes with experimental gingivitis. Huang et al. (2014) demonstrated a decrease in relative abundance of 5 bacterial genera, including Streptococcus, between baseline and 3-wk experimental gingivitis and an increase in 22 bacterial genera, including Prevotella and Selenomonas. Similarly, Kistler et al. (2013) identified members of the Streptococcus, Rothia, and Actinomyces genera as decreasing in relative abundance after 2 wk of experimental gingivitis, whereas the genera Campylobacter and Selenomonas showed increases in relative abundance as gingivitis developed.
This study was designed to understand the global changes in microbiome diversity and composition in the supragingival biofilm transcriptome associated with fixed appliances in an adolescent population. Importantly, this analysis revealed possible bacterial biomarkers of oral disease risk, taking a first step toward pretreatment screening. S. longum, M. diversum, and N. subflava in saliva were identified as potential biomarker species that could be measured at baseline to predict later oral health status during treatment (Fig 4D). Baseline abundance of S. longum and M. diversum were elevated in subjects who displayed increased levels of BOP at 12 wk postbonding. S. longum is implicated in ECC and worsened periodontal health in young adults, consistent with this species serving as a candidate biomarker of inflammatory risk and playing a potential role in a health-to-disease transition (Islam et al. 2020; Cho et al. 2023). M. diversum is a bacterial strain of anaerobic gram-positive rods and has been implicated in the development of chronic periodontitis (Nakazawa et al. 2000). N. subflava was found to be higher in the baseline saliva of subjects with better gingival health (<40% sites with BOP) at 12 wk postbonding. N. subflava is a commensal bacterium, and its higher abundance was associated with periodontal health (Lenartova et al. 2021).
The identification of potential salivary microbial biomarkers associated with an increased risk of gingival inflammation from orthodontics reinforces the value of saliva tests for personalized care in dentistry. Patients could provide saliva at initial consultation to render a risk profile for developing caries and gingivitis during treatment based on the presence of biomarker species; results could guide increased hygiene efforts, higher frequency of oral prophylaxis, and potentially accelerated treatment timing for patients with harbingers of disease.
Limitations of this study include a small sample and lack of full clinical data for all time points due to the COVID-19 pandemic and other factors related to attendance. Future directions include longer follow-up with a larger sample. A larger sample may allow for a comprehensive study of the impact of demographics (gender, age, and race) along with identification of pathogens responsible for periodontal conditions observed during orthodontics, including gingival hyperplasia. Longer follow-up would allow for development and detection of enamel decalcification, appearing as white spots and cavitated lesions. It is notable that gingival index and BOP data appear to cluster into 2 groups that may be indicative of oral states of health and disease warranting further investigation with an adequately powered sample (Fig. 2A, B).
The present study used RNA sequencing to study changes in the supragingival biofilm transcriptome before and after bonding of fixed appliances. RNA-seq did not directly quantify bacterial abundance; instead, it provided measures of relative abundance of species that are present in the supragingival biofilm and remain biologically or transcriptionally active (i.e., generating RNA) during the study period. These species-level RNA-seq data reflect a dysbiotic shift in communities with observed changes in microbial candidates parallel to what is expected in a disease-associated dysbiotic shift. In summary, results suggest that the introduction of fixed appliances could serve as a clinical research model, enabling investigators to test preventive treatment modalities in vivo. Future studies can leverage this RNA-seq data by investigating specific bacterial genes’ upregulation and downregulation, as well as associated gene sets and biological pathways at play during orthodontic treatment. Such information can be relevant to understanding early stages of periodontal inflammation and caries development and may help identify predictive oral disease biomarkers, as well as candidate targets for intervention.
Conclusions
Results provide proof-of-principle that an elective oral disease risk-increasing intervention, fixed orthodontic appliances, can induce a dysbiotic shift in the supragingival oral biofilm of healthy patients that parallels clinically evident changes in periodontal health. These data reveal that pathogenic and homeostatic species vary significantly after bonding and may contribute to the development of oral dysbiosis and clinical disease. Future studies must delve into the role of such species with a larger sample and longer-term follow-up, to explore the bacterial pathogenesis of gingival inflammation and caries in a healthy population.
Author Contributions
E. Babikow, N. Ghaltakhchyan, T. Livingston, Y. Qu, C. Liu, J. Roach, D. Wu, L. A. Jacox, contributed to data analysis, drafted the manuscript and critically revised the manuscript; A. Hoxie, T. Sulkowski, C. Bocklage, contributed to data analysis, drafted the manuscript; A. Marsh, K. B. Mitchell, A. De A. Ribeiro, contributed to data analysis, critically revised the manuscript; S. T. Phillips, T. H. Jackson, contributed to conception, design, critically revised the manuscript; K. Divaris, contributed to conception, design, data analysis, drafted the manuscript, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jct-10.1177_23800844231199393 – Supplemental material for Longitudinal Microbiome Changes in Supragingival Biofilm Transcriptomes Induced by Orthodontics
Supplemental material, sj-docx-1-jct-10.1177_23800844231199393 for Longitudinal Microbiome Changes in Supragingival Biofilm Transcriptomes Induced by Orthodontics by E. Babikow, N. Ghaltakhchyan, T. Livingston, Y. Qu, C. Liu, A. Hoxie, T. Sulkowski, C. Bocklage, A. Marsh, S. T. Phillips, K. B. Mitchell, A. De A. Ribeiro, T. H. Jackson, J. Roach, D. Wu, K. Divaris and L. A. Jacox in JDR Clinical & Translational Research
Footnotes
Acknowledgements
We thank our participants for their time and engagement with this study. We appreciate the UNC Department of Orthodontics, its faculty and residents, for hosting this investigation. We thank Dr. Ashley Morgenstern, DDS, MS, for helping to initiate this project. We are grateful for the excellent UNC Microbiome core, in particular Andrea Azcarate-Peril and Al-Mounawara Abiodoun Yaya for their expertise and support. We appreciate the support and guidance of the UNC Adams School of Dentistry GoHealth Clinical Research Unit (CRU).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Southern Association of Orthodontists Research Award (to T.L.) and the American Association of Orthodontists Foundation (AAOF) Orthodontic Faculty Development Fellowship Award (to T.J.), the Research Aid Awards (to E.B.), and the Martin “Bud” Schulman Postdoctoral Fellowship Award (to L.J.). The project was funded by the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH), through Grant Award UL1TR002489 (to T.J.) and by the National Institutes of Dental and Craniofacial Research (NIDCR), NIH through a K08 award (to L.J.), with Grant Award 1K08DE030235-01A1. This work was supported by research grants from the NIH: NIDCR U01DE025046 (K.D.) and R03DE028983 (D.W.). The UNC Microbiome Core Facility is supported by the Center for Gastrointestinal Biology and Disease (CGIBD P30 DK034987) and the UNC Nutrition Obesity Research Center (NORC P30 DK056350). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Data Availability Statement
The data supporting the findings of this study are available within the article and its supplementary materials.
A supplemental appendix to this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.