
Abstract
Fibrous dysplasia is a rare non-hereditary congenital condition characterized by 2 main forms: monostotic and polyostotic. Monostotic is the more common form, while polyostotic, often associated with a syndrome, is rarer. The case presented involves a 10-year-old patient who was diagnosed with polyostotic fibrous dysplasia accompanied by an endocrinopathy. This report explores the clinical and radiological aspects of this condition based on the patient’s case.
Introduction
Fibrous dysplasia is a rare congenital bone disease that is non-hereditary and is secondary to an activating mutation in the GNAS1 gene (Guanine Nucleotide Binding Protein, alpha stimulatory). 1 Morphologically, it is characterized by the replacement of spongy bone with immature bone that is low in trabeculae and rich in fibrous connective tissue. Two forms have been described, the more common monostotic form that affects a single bone, and the less common polyostotic form. Fibrous dysplasia is often asymptomatic and is discovered incidentally or revealed by a complication. The polyostotic form is often associated with a syndrome. 2
Case Report
This is a 10-year-old child with good psychomotor development, no consanguinity in the family, and a history of epilepsy under treatment. She has been experiencing walking difficulties and left hip pain for the past year. Clinical examination revealed a limp while walking and pain upon mobilization of the left hip. Given these clinical signs, clinicians requested standard pelvic radiographs and MRI of the lower limbs to investigate signs of proximal femoral focal deficiency (PFFD). The results of the radiographs and MRI ruled out PFFD. The standard radiograph showed mixed sclerotic and lytic lesions with thinning of the cortical bone without rupture (Figure 1). The MRI of the lower limbs revealed bilateral metaphyseal-diaphyseal femoral signal abnormalities, with hypointensity on T1, intermediate signal on T2, heterogeneous enhancement on STIR after contrast injection, and signal abnormalities in the femoral neck, with hypointensity on T1, hyperintensity on T2, and annular enhancement after contrast injection (Figures 2 and 3). Fibrous dysplasia was suggested based on these radiological signs. Our clinical examination also identified right exophthalmos and right frontal depression without café-au-lait spots. The rest of the clinical examination was normal. A brain CT scan was performed to explore the cause of exophthalmos and facial asymmetry. The brain CT scan showed a characteristic ground-glass appearance of bone dysplasia associated with exophthalmos (Figure 4). Abdominal and cervical ultrasound examinations were conducted as part of the search for associated endocrinopathies, revealing lesions suggestive of thyroiditis (Figure 6). A brain MRI was performed as part of the epilepsy investigation, which showed thickening of the right frontal and occipital bones, as well as the bones of the facial skeleton, along with exophthalmos and meningeal enhancement (Figure 5). The patient was referred to endocrinology, ophthalmology, otolaryngology (ENT), and maxillofacial surgery for ongoing management of her condition.

Pelvic radiography (Anterior and lateral views) and right leg radiography (Anterior and lateral views) showing mixed lytic and sclerotic lesions(arrows) with cortical involvement(star).
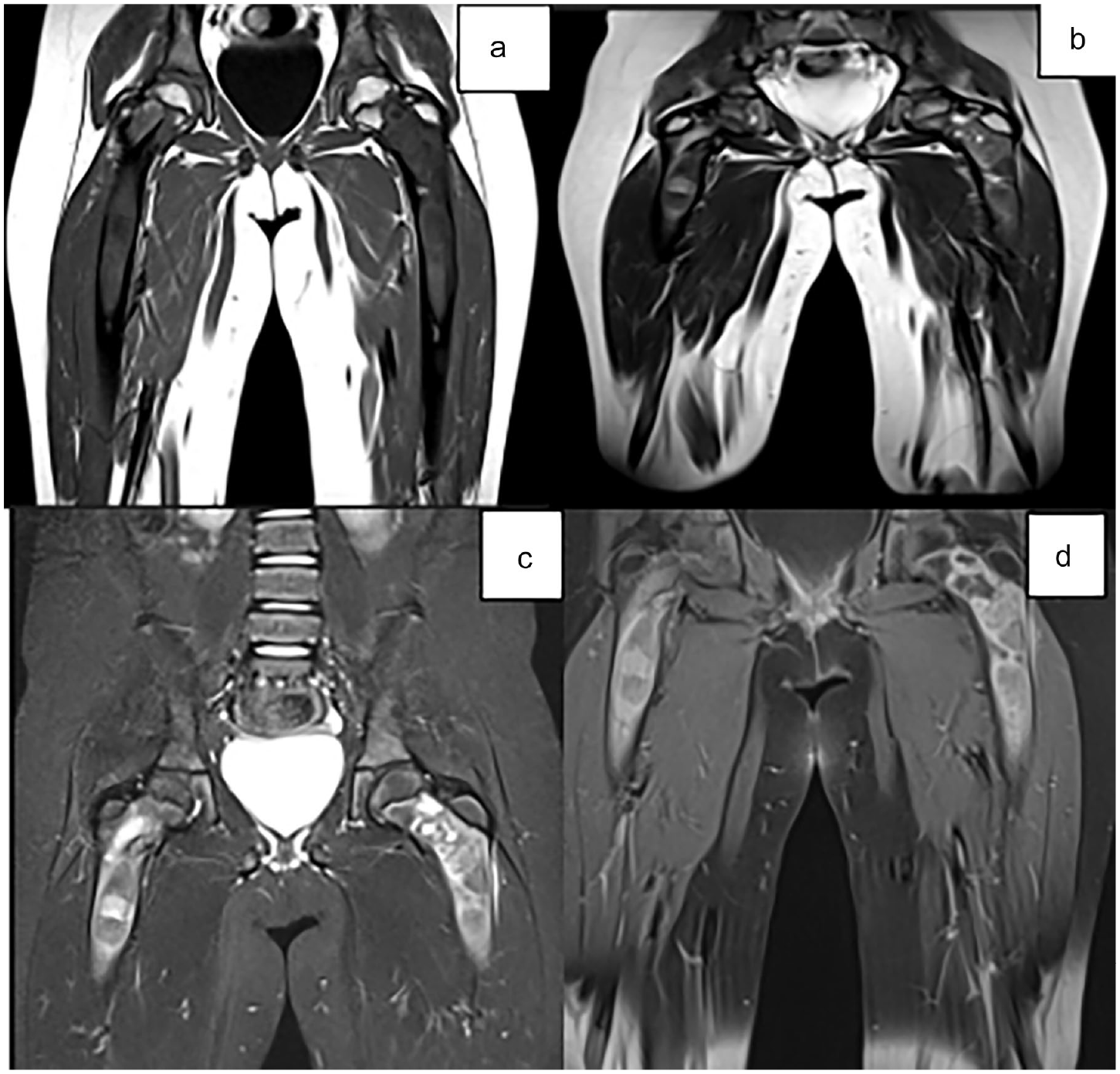
Coronal sections of an MRI of the lower limbs showing bilateral femoral metaphyseal-diaphyseal signal abnormalities, with T1 hypointensity (a), intermediate signal on T2 (b), STIR hyperintensity (c), enhanced heterogeneously after contrast injection (d), blowing the cortex without breaking it.

Coronal sections encompassing the knees and legs showing signal abnormalities in the right tibial region with T1 hypointensity (a), T2 hyperintensity on STIR (b), enhanced after contrast injection (c).
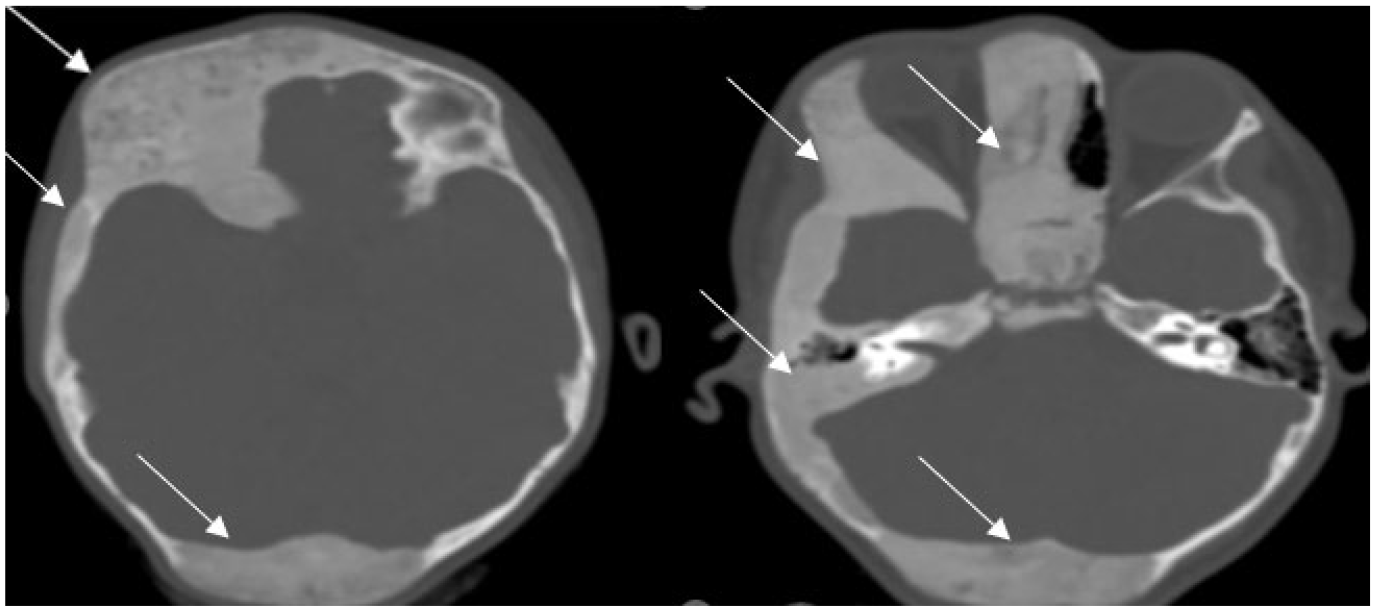
Axial sections of a brain CT scan showing thickening with a ground glass appearance of the right frontal, temporal, and occipital bones(arrows), as well as the entire right facial structure, with grade 1 exophthalmos.

Sagittal and axial sections showing bone thickening of the right frontal, occipital, and right facial bones with iso-signal on T1 (a and b), heterogeneous hyperintensity on T2 Flair (c), enhancing heterogeneously after contrast injection with enhancement (d).

Axial and sagittal sections of a cervical ultrasound revealing a heterogeneous pseudo-nodular thyroid with hypervascularity on color Doppler.
Our patient was placed on valproic acid to prevent seizure episodes and received symptomatic pain treatment with acetaminophen, showing good clinical improvement and under multidisciplinary medical supervision by an orthopedic pediatric surgeon, endocrinologist, ophthalmologist, maxillofacial surgeon, pediatric neurologist, and radiologist.
Discussion
Fibrous dysplasia (FD) is an idiopathic and benign bone dystrophy. Histologically, it is characterized by the replacement of the medullary cavity of bones with fibro-osseous tissue, resulting in distortion and hypertrophy of the affected bone. 3
Fibrous dysplasia is a congenital disease caused by a mutation in the GNAS 1 gene. It involves an activating mutation of the alpha subunit of the G protein, leading to increased adenylyl cyclase activity, overproduction of cAMP, and subsequent overexpression of the c-fos protein, resulting in a defect in osteoblast differentiation. 4
The prevalence of fibrous dysplasia is unknown, but it is estimated to be around 1 in 30,000 individuals. It represents 2.5% of bone diseases and 7% of bone tumors. Fibrous dysplasia of the bones can affect both sexes, with a sex ratio of 1. The age at diagnosis is typically between 5 and 30 years old.
Our patient was female and she was 10 years old, which is the average age of disease diagnosis according to the literature.
In most cases, bone lesions show minimal progression after puberty. The risk of malignant transformation is estimated to be between 0.5% and 4% in different series. Fibrous dysplasia can affect any bone in the body. The involvement can be monostotic or polyostotic. In the monostotic form, which accounts for 70% to 80% of cases, the most commonly affected bones are the ribs (45%), the craniofacial bones including the maxilla and cranial vault, and the proximal femur. In long bones, the typical involvement is in the metaphyseal-diaphyseal region. In the polyostotic form, there is often a unilateral or predominantly unilateral distribution of affected bone sites. 3
In our patient, craniofacial involvement was predominant on the right side. The involvement of the lower limbs affected both limbs.
Associations: Although fibrous dysplasia is generally sporadic several associations are well-recognized:3,5
McCune-Albright syndrome: in 2% to 3% of cases with the polyostotic form. 6 This syndrome encompasses polyostotic fibrous dysplasia, endocrinopathy (early puberty), and café-au-lait spots. 7
Mazabraud syndrome: myxomas of soft tissues (rare); typically multiple intramuscular lesions near the most severely affected bone.
Isolated endocrinopathy without complete early puberty of McCune-Albright syndrome in girls:
hyperthyroidism, hyperparathyroidism: kidney stones, calcinosis, acromegaly, diabetes mellitus, Cushing’s syndrome: osteoporosis, acne, and growth delay.
In our patient, she had polyostotic bone dysplasia, sonographic signs of thyroiditis with euthyroidism, and meningeal enhancement in relation to cranial lesions likely causing her seizures.
Fibrous dysplasia of the bones is often asymptomatic and is typically discovered incidentally on standard X-rays taken for another reason. It can be revealed by pain, which can originate from the bone or joint when secondary osteoarthritis is present. The pain often results from a crack, pathological fracture, or bone deformation with functional impairment. Deformities associated with fibrous dysplasia include a shepherd’s crook deformity of the tibia, femur, or humerus, genu varum or genu valgum, and unequal length of the lower limbs. Involvement of the skull base can manifest as facial asymmetry, exophthalmos, and abnormalities in dental joint development. 8
In our patient, the involvement of the lower limbs resulted in a limp. The craniofacial involvement led to exophthalmos and facial asymmetry.
The diagnosis is most often confirmed by radiological findings. These findings typically reveal an osteolytic lesion, often heterogeneous, predominantly radiolucent, with thinning of the cortical bone, sometimes bone hypertrophy, and often a peripheral rim of osteosclerosis around the lesion. However, in some areas, there is a bone density known as “ground glass” which is highly suggestive of the diagnosis. 9
The preferred sites include the diaphysis of long bones, metaphyses, especially the femoral neck, ribs, spine, and craniofacial region. CT scans allow for the assessment of fracture risk by measuring cortical thickness and identifying fissures not visible on standard X-rays. Bone scintigraphy is performed at the beginning of management to detect all bone lesions of FD; dysplastic lesions are hyperfixative. MRI has no role in the diagnosis of FD but may be useful if there is suspicion of malignant transformation.
The treatment is primarily symptomatic. Pain management follows the World Health Organization’s (WHO) analgesic ladder, including non-steroidal anti-inflammatory drugs (NSAIDs) and medications aimed at neuropathic pain, such as amitriptyline and pregabalin. Occasionally, a short course of corticosteroids at a dose of 1 mg/kg/day may be necessary, especially in cases of nerve compression syndrome, including optic nerve compression. 2 Some recent publications have suggested the significant benefits of bisphosphonates. Indications for bisphosphonates: Muscular pain resistant to symptomatic treatment, usual analgesics, or NSAIDs; destabilizing lesions at risk of fracture without surgical indication and associated with elevated bone remodeling markers; effectiveness will be assessed using a pain scale (VAS), bone markers, and radiographic imaging. Surgical treatment is indicated in cases of nerve compression for excision or fracture. 2
Conclusion
Fibrous dysplasia is a rare benign disease. Imaging now greatly facilitates the diagnosis of this condition. It is most often asymptomatic and discovered incidentally. However, in rare cases, there may be bone pain, deformity, or it may be revealed by complications such as pathological fractures, nerve compression, or extremely rare sarcomatous transformation. The treatment is essentially symptomatic.
Footnotes
Acknowledgements
I would like to express my gratitude to my professors and all the colleagues who participated in the completion of this work.
Author Contributions
IH: Contributed to conception and design; Contributed to analysis; Drafted the manuscript; Gave final approval; Agrees to be accountable for all aspects of work ensuring integrity and accuracy. SE, SO and FC: Contributed to conception and design; Contributed to analysis; Agrees to be accountable for all aspects of work ensuring integrity and accuracy. NB: Critically revised the manuscript; Gave final approval; Agrees to be accountable for all aspects of work ensuring integrity and accuracy. YG: Contributed to conception and design; Contributed to analysis; Agrees to be accountable for all aspects of work ensuring integrity and accuracy. NA, LC and SEH: critically revised the manuscript; Gave final approval; Agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Written informed consent was obtained from the parents of patients for the publication of this case report.
Guarantor of Submission
The corresponding author is the guarantor of submission