
Abstract

Introduction
Pseudohypoaldosteronism type II (PHA II), which is also known as Gordon syndrome, is a rare autosomal dominant disease characterized by hypertension, hyperkalemia, hyperchloremic metabolic acidosis, and the absence of hyponatremia and hyperaldosteronemia. 1 The pathophysiology of PHA II is explained by the constitutive activation of WNK (with no lysine) kinases–OSR1/SPAK kinases–sodium/chloride cotransporter (NCC) phosphorylation cascade. NCC is distributed on the apical plasma membranes of the distal convoluted tubules, and activation of NCC mediates increasing sodium chloride (NaCl) reabsorption in the kidney, leading to hypertension, hyperkalemia, and metabolic acidosis. 2 Mutations of 4 genes, WNK1, WNK4, KLHL3 (Kelch-like 3), and CUL3 (Cullin 3), have been reported to be responsible to PHA II.3,4
Here, we present the case of a 6-year-old Japanese boy with PHA II. Our case is the third one with novel mutation in CUL3 gene. We also studied the excretion of urinary total NCC (tNCC) and phosphorylated NCC (pNCC) in his family members.
Case Report
The patient was a 6-year-old boy and the second child of nonconsanguineous healthy Japanese parents. He was delivered at 40 weeks and 3 days of gestation by cesarean section, because his mother was a woman with previous cesarean birth. His birth weight was 2748 g (−1.6 standard deviation [SD] in Japan), and his body length was 46.0 cm (−2.0 SD). At the age of 2 years, he had a mild language disorder and was monitored by a pediatric neurologist at our hospital. Since the age of 3 years, he required dental care for his decayed carious teeth. For the induction and maintenance of anesthesia, rocuronium bromide, propofol, remifentanil hydrochloride, nitrous oxide, and sevoflurane were used. During the dental operation, blood data showed hyperkalemia (9.43 mmol/L) and metabolic acidosis (pH 7.24; serum bicarbonate level, 16.8 mmol/L; base excess, −9.9 mmol/L, anion gap 12.9). Fortunately, the patient recovered fully from anesthesia without any treatment.
At the age of 6 years, he was admitted to our hospital for investigation of the causes of hyperkalemia and metabolic acidosis. Those were discovered incidentally, when he was screened for short stature. His height was 104.0 cm (−2.3 SD), and his body weight was 17.2 kg, which was 103% of the Japanese standard weight for his height. The patient’s blood pressure was high, at 130/88 mmHg. The physical findings were normal, except for severely decayed teeth. The findings of our patient are summarized in Table 1. Blood examination showed metabolic acidosis and electrolyte imbalance. This serum alkaline phosphatase level was elevated to 1954 IU/L. Serum levels of calcium, phosphorus, and parathyroid hormone and the liver function were normal. The levels of insulin-like growth factor-1 (IGF-1) and thyroid hormones were also normal. The bone age evaluated by the Tanner-Whitehouse 2 method was delayed by approximately 2 years related to his chronological age. No abnormalities were detected on chest radiography and magnetic resonance imaging of the brain. Electrocardiogram revealed tall peaked T waves, presumably due to hyperkalemia. Ultrasound study of the heart and kidneys revealed no abnormalities. Laboratory studies for renal function showed the following results: serum urea nitrogen level, 13.1 mg/dL; serum creatinine level, 0.35 mg/dL; serum cystatin C level, 0.57 mg/L; creatinine clearance, 231.2 to 235.0 mL·min−1·1.73 m−2. These findings helped rule out renal insufficiency, which is a common cause of hypertension, hyperkalemia, and metabolic acidosis. Fractional excretion of potassium (FEK) was 2% (normal = 4% to 10%), and transtubular potassium gradient (TTKG) was low at 4.14 (normal = 7-10) despite hyperkalemia. The NCC activity was measured by immunoreactivity assay reported previously. 5 The excretion of urinary tNCC was 236- and 56.2-fold higher than those of his father and elder sister, respectively. The excretion of pNCC was 8.6-, 11.6-, and 14.1-fold higher than those of his father, mother, and elder sister, respectively (Table 1). The furosemide upright test and rapid adrenocorticotropic hormone (ACTH) test revealed that he had a normal renin–angiotensin–aldosterone system and adrenal cortical function. Dietary salt restriction (3 g/day) for 5 days improved his electrolyte levels and acid–base balance (serum potassium level, 5.9 mmol/L; serum chloride level, 111 mmol/L; pH 7.337; serum bicarbonate level, 18.6 mmol/L; base excess, −7.2 mmol/L). The administration of hydrochlorothiazide (0.05-0.07 mg/kg daily) for 1 week further improved these parameters (serum potassium level, 5.1 mmol/L; pH 7.389; serum bicarbonate level, 22.2 mmol/L), in addition to the FEK (from 2.0% to 6.3%), plasma rennin activity (from 0.6 to 11.4 ng·mL−1·h−1), plasma aldosterone concentration (from 420 to 785 pg/mL), and blood pressure (from 130/88 to 101/70 mm Hg). At the time of the last follow-up, his height and bone age gradually improved. His height was −2.1 SD and bone age was delayed by about 1 year.
Features of Pseudohypoaldosteronism Type II Patients With CUL3 Gene Mutation.
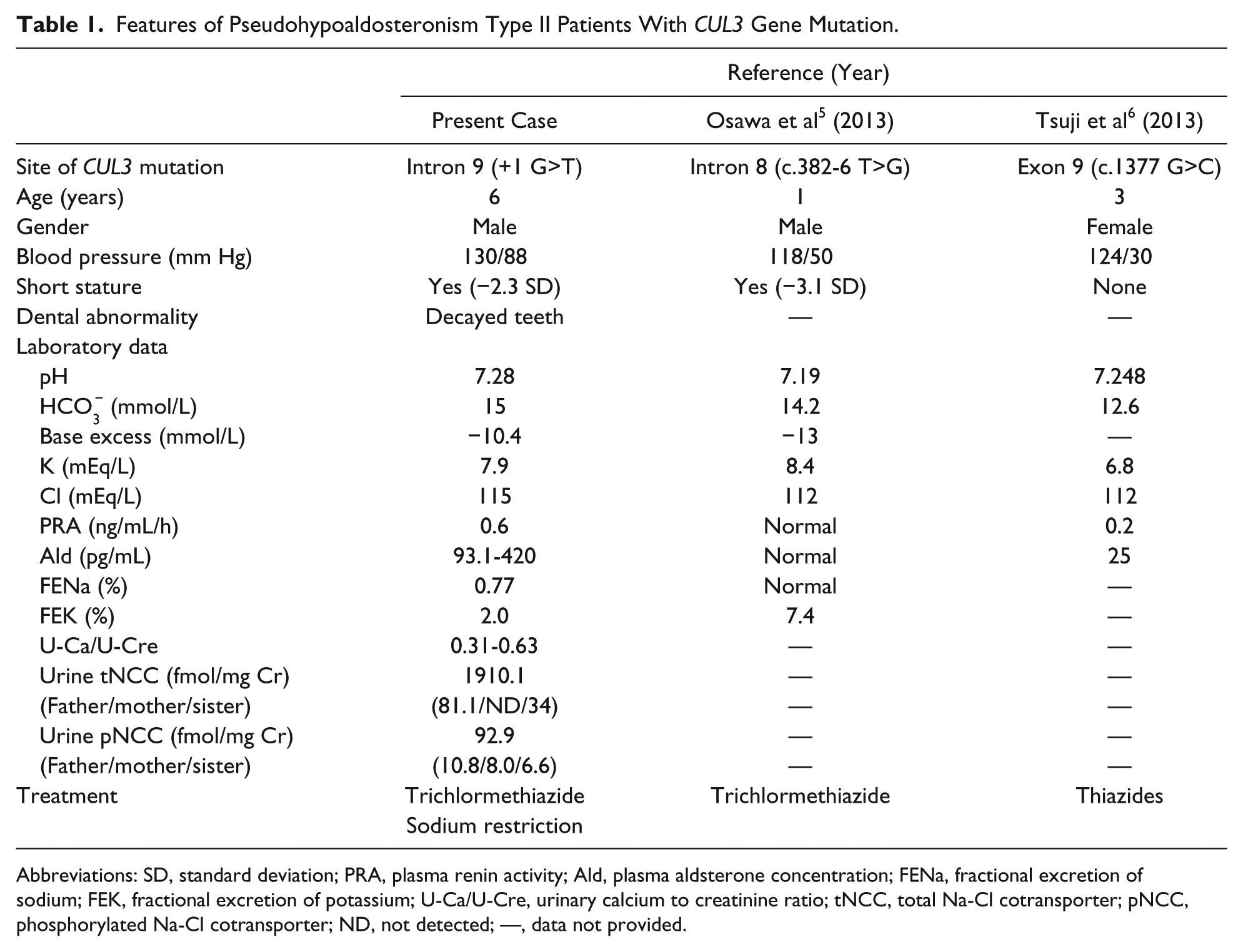
Abbreviations: SD, standard deviation; PRA, plasma renin activity; Ald, plasma aldsterone concentration; FENa, fractional excretion of sodium; FEK, fractional excretion of potassium; U-Ca/U-Cre, urinary calcium to creatinine ratio; tNCC, total Na-Cl cotransporter; pNCC, phosphorylated Na-Cl cotransporter; ND, not detected; —, data not provided.
Gene analyses were performed after obtaining informed consent. As shown in Figure 1a, DNA sequencing revealed a mutation in CUL3 intron 9; no such deletion was observed in his parents. The reverse transcription-polymerase chain reaction (RT-PCR) assay showed deletion of exon 9 in CUL3 mRNA of his white blood cells. Exon 9 skipping was confirmed by direct sequencing of the PCR-amplified fragment. A heterozygous nucleotide substitution was identified in the splice-accepter site of the CUL3 intron 9 (+1 G>T). In some of the reported cases of PHA II due to mutation in CUL3, autosomal dominance was detected; therefore, the DNA of his parents was analyzed. However, no such mutation was seen in samples from either of his parents, and we concluded that the mutation in the CUL3 gene was de novo (Figure 1b).

Gene analyses in our patient.
Discussion
We report the case of a 6-year-old Japanese boy with PHA II caused by a mutation in CUL3 intron 9 (+1 G>T), resulting in the skipping of exon 9. We also showed here activated NCC function by measuring the urinary tNCC and pNCC protein levels in our patient.
Compared with PHA II patients with WNK1, WNK4, or KLHL3 mutations, patients with CUL3 mutations tend to have more severe phenotypes at a younger age than those with other mutations; furthermore, the frequency of de novo mutations is higher in CUL3 than that in other genes.3,5 Two cases of PHA II due to CUL3 mutation have been reported thus far (Table 1).5,6 Including our patient, 2 of the 3 patients showed growth retardation and all had hyperkalemia, hyperchloremia, metabolic acidosis, and hypertension.
Recently, sensitive and quantitative assays for urinary NCC have been established and high levels of excretion of tNCC and pNCC in PHA II patients have been found. 7 Measuring the urinary pNCC levels was useful also in our patient clearly showing activated NCC in PHA II by CUL3 mutation.
Boyden et al conducted a genetic analysis of 17 PHA II families with the CUL3 mutation and reported that the splice acceptor is intron 8, which is responsible for the skipping of exon 9 in all their patients. 3 Similar to their reports, our patient’s CUL3 mRNA was aberrantly spliced inducing skipping of exon 9 of CUL3 mRNA. However, the impaired splicing was induced by a novel single-nucleotide substitution in the splice-acceptor site of CUL3 intron 9.
Pediatric patients with PHA II occasionally develop decayed carious teeth and require dental care under general anesthesia. 8 The drugs for anesthesia, particularly succinylcholine, can aggravate hyperkalemia under several conditions, including PHA II.8,9 Therefore, we should pay attention to hyperkalemia induced by succinylcholine when anesthesia is performed in PHA II patients. It is unclear why severe hyperkalemia did not induce any cardiac complications, such as ventricular tachycardia with critical hypotension, in our patient.
Acidemia in PHA II patients is considered to be a consequence of hyperkalemia. 1 It is known that chronic metabolic acidosis inhibits growth because it adversely affects bone metabolism, skeletal growth, and sensitivity to growth hormone (GH) and IGF-1. 10 Our patient had short stature but showed normal levels of IGF-1 (−0.6 SD for his age). His urinary calcium excretion was high and his bone density was low, and his bone age was 2 years less than his chronological age. These indicate that the short stature in our patient was due to the impaired bone metabolism. By the treatment with hydrochlorothiazide, which inhibits NCC function, hyperchloremia, hyperkalemia, metabolic acidosis, hypertension, and urinary calcium-to-creatinine ratio were improved in our patient, indicating that patients with CUL3 mutations are not resistant to this drug, even if the symptoms are severe.
In conclusion, early diagnosis and intervention could be beneficial for pediatric PHA II patients and improve the prognosis.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by a Grant-in-Aid for Scientific Research (C) #24591525 from the Ministry of Education, Culture, Sports, Science and Technology, Japan.