
Abstract
A 63-year-old woman with common variable immunodeficiency (CVID) presented with 1 year of insidious onset lower extremity pain and weakness. She underwent a circuitous workup, failed to improve despite treatment for various presumed diagnoses. She presented to a University physical medicine and rehabilitation clinic with continued symptoms. Electrophysiologic testing was recommended revealing a lower extremity motor greater than sensory axonal neuropathy. While CVID has known central nervous system complications, to our knowledge, this represents the second known reported case of peripheral neuropathy. We review the literature on CVID and summarize neurological disease mechanisms and manifestations. Although peripheral neuropathy is a rarely documented complication of CVID, providers need to be aware of potential peripheral nervous system complications of primary immune deficiencies such as CVID due to its significant impact on physical performance, balance, and fall risks.
Keywords
Introduction
Neurological symptoms are a recognized feature of various immunodeficiency diseases; primary (Notarangelo et al., 2004) and combined immunodeficiencies (Al-Herz et al., 2011) can have neurological manifestations. In combined immunodeficiency disease, neurological symptoms develop due to elevated levels of toxic metabolites and dysregulation of apoptosis (Al-Herz et al., 2011). Neurological complications, however, are less commonly documented as a feature of primary antibody deficiencies such as common variable immunodeficiency (CVID; Hermaszewski & Webster, 1993). Although a few cases are reported, most are cases of central nervous system (CNS) disease including myelopathy, encephalopathy, or encephalomyelitis (Kondo et al., 2005; Rudge et al., 1996). The incidence of peripheral nerve disease due to CVID is not known. The pathophysiology of CNS complications in antibody deficiencies such as CVID is unclear, but a number of primary antibody deficiency known features have been proposed to play a role. These include immune dysregulation, inflammatory processes, and susceptibility to rare infections (Snider et al., 1983). Additional theories propose a link between intravenous immunoglobulin (IVIg) therapy and onset of neurologic symptoms, which may progress into neurodegenerative disorders (Ziegner et al., 2002). Even more rare, however, are cases in which CVID patients present with symptoms of peripheral neuropathy. Reported only once previously in the literature, we introduce an unusual presentation of a case of peripheral neuropathy in a patient with CVID (Larner, Webster, & Thomas, 2000). We summarize the clinical features, review the literature, and provide clinical commentary on making the diagnosis, alerting health care providers to this unique complication to permit an earlier workup and prompt diagnosis.
Method
Case Description
A 63-year-old woman with CVID presented in July 2014 with a 1 year of insidious onset distal lower extremity discomfort and weakness, worsening over 3 months. Her past medical history was remarkable for asthma requiring chronic corticosteroids leading to adrenal insufficiency, osteoporosis, depression, insomnia, migraines, and orthopedic complaints including right knee meniscal tear, left knee osteoarthritis, left Achilles tendon tear, and right ankle osteoarthritis. Her CVID was diagnosed prior to initial evaluation at our facility; diagnosis was based on Pan American Group for Immune deficiency (PAGID) and European Society for Immunodeficiencies (ESID) criteria based on available records. She had no thyroid disease, diabetes, chronic liver or kidney disease, cancer/paraneoplastic syndrome, monoclonal gammopathy, chronic infection (e.g., lyme, HIV), vitamin B6/B12 or other nutritional deficiencies, alcoholism or other toxin exposures, or other common causes of peripheral polyneuropathy (Azhary, Farooq, Bhanushali, Majid, & Kassab, 2010). Past surgical history included left Achilles tendon repair, right knee arthroscopy, and right total knee arthroplasty. She denied smoking, tobacco, or other drug use. She noted a family history of breast cancer. Medications included prednisone (10 mg once a day), omalizumab, fluticasone-salmeterol inhaler, fludrocortisone, ibandronate, and bupropion. She began IVIg replacement therapy (10% human,10 g/100 mL IV injection) monthly for CVID since 2009 (historical data on IgG levels before IVIg were unavailable).
Due to right ankle pain and stiffness, she underwent total ankle arthroplasty in October 2015. She did not experience any neurologic injury due to arthroplasty. Following the procedure, her generalized weakness and pain symptoms persisted. In March 2016, she underwent rheumatologic workup for her diffuse symptoms, which was ultimately negative. Her physician at the time proposed type 1 complex regional pain syndrome. In July 2016, she underwent lumbar spine magnetic resonance imaging (MRI) for back pain, which showed multilevel degenerative changes at L3-4, L4-5, L5-S1 with moderate bilateral neural foraminal and central canal stenosis expected for age, but no nerve root compression or displacement of cauda equina nerve roots. Epidural steroid injections provided only temporary relief from back pain but not her foot symptoms.
In January 2017, she presented to an academic Physical Medicine & Rehabilitation clinic. At this point, she had additionally seen a physical therapist, a chiropractor, and a podiatrist without improvement. Upon closer review, her back pain subsided but she was reporting worsening bilateral foot numbness. Further review of systems was unremarkable. On examination, height was 5′3″ (1.6 m), weight 50.3 kg, with body mass index (BMI) 19.7. Gross lower extremity sensation was intact, but she felt decreased subjective sensibility in the plantar soles and metatarsal head regions. Lumbar and lower extremity range of motion and gross motor strength were within normal limits for age, no muscle stretch reflex asymmetry, or pathologic reflexes. She had no lower extremity edema. Provocative testing including straight leg raise was negative. Gait showed mild increase in double support time, cautious gait but minimal unsteadiness.
Recent blood testing was within normal limits, including glucose (87 mg/dL), HbA1c (5.4%), Vitamin B12 (745 pg/mL), thyroid function tests (thyroid stimulating hormone [TSH] = 1.72 uIU/mL, free T4 = 1.13 ng/dL), erythrocyte sedimentation rate (18 mm/h), and C-reactive protein (0.2 mg/dL). Other tests included normal blood urea nitrogen (BUN)/creatinine 18/0.94 mg/dL, glomerular filtration rate (>60 mL/min), aspartate transaminase (AST)/alanine aminotransferase (ALT) 8/26 U/L, white blood cells (WBC) 4.9, hemoglobin 12.9. Rheumatoid factor (<10 IU/mL), cyclic citrullinated peptide antibody IgG (<8 U/mL), creatinine phosphokinase (91 U/L) were within normal limits and anti-nuclear antibody was negative. Immunoglobulin levels showed low IgG subclass 3 (19 mg/dL); other IgG subclasses 1, 2, and 4, IgA, and IgM were within normal range.
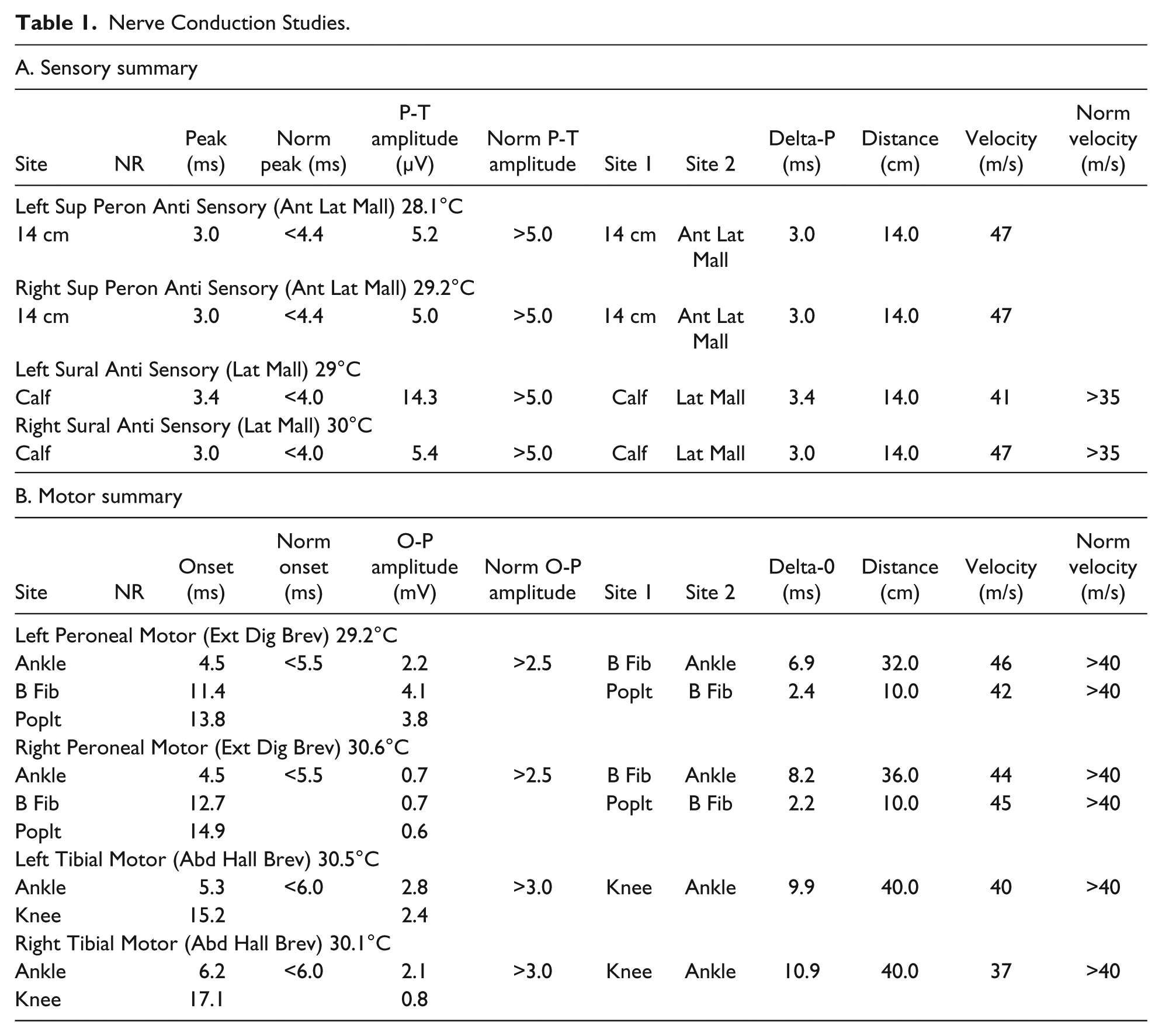
Nerve conduction studies (NCS) and electromyography (EMG) of bilateral lower extremities were ordered for further assessment. In February 2017, she underwent NCS and EMG testing (see Table 1 and Table 2 for results, respectively). She did not want extensive testing due to discomfort; F waves, H reflex, and upper extremity testing therefore were not done. She had no back pain and declined expanded EMG including lumbar paraspinals. Relevant findings included reduced bilateral tibial motor (right = 2.1 mV, left = 2.8 mV, lab norm > 3.0 mV) and right peroneal motor amplitudes (right = 0.7 mV, left = 2.2 mV, lab norm > 2.5 mV; possible accessory left deep peroneal nerve noted). There was borderline prolonged distal latency (6.2 ms, lab norm = 6.1 ms) and conduction velocity (37 m/s, lab norm = 38 m/s) of the right tibial motor nerve; with temperature correction to 32°C (2 m/s/1°C, 0.2 ms/1°C; Halar, DeLisa, & Brozovich, 1981; Halar, DeLisa, & Soine, 1983), this was considered normal at 5.8 ms and 40 m/s. There were borderline reduced right sural (5.4 uV) and bilateral superficial peroneal sensory amplitudes (right = 5.0 uV, left = 5.2 uV), but as colder limb temperatures cause artificially high amplitudes (Morris, 2013; Moses, Nelson, Nelson, & Cheifetz, 2007; Nelson et al., 2004; Rutkove, 2001), the true motor and sensory amplitudes are considered further reduced. EMG demonstrated mild chronic neurogenic changes bilateral S1 and left L5 myotomes. Other than low-amplitude positive sharp waves in right medial gastrocnemius, there was no evidence of denervation. While chronic neurogenic changes would be compatible with an axonal neuropathy, radiculopathies could not be excluded. Bilateral multilevel radiculopathies could be consistent with lumbar spinal stenosis; however, clinically she was not reporting back pain with radicular symptoms or other symptoms consistent with neurogenic claudication, and lumbar paraspinals are only positive in 46% of cases (Hall et al., 1985). Paraspinal EMG was declined as mentioned, but paraspinal responses are variable and can be of limited value (Barr, 2013; can be normal with radiculopathy, or abnormal without radiculopathy, for example, in older patients, diabetics, etc.). Therefore, the constellation of findings would suggest a distal symmetric lower extremity sensorimotor neuropathy, not severe enough to generate significant positive EMG findings.
Nerve Conduction Studies.
Electromyography.

The patient was referred back to her primary team for further management. In July 2017, her CVID treatment was switched to subcutaneous IgG (human, 20% SQ) injection every 2 weeks and she gradually felt improvement. At last exam, September 2018, she was doing well with less lower extremity complaints. It is unknown but postulated the adjustment in CVID treatment helped her neuropathy symptoms. She did not receive any other treatments for neuropathy, for example, neuropathic medication or immunotherapy (patient was treated outside our institution and further data on primary treating team’s reason for change unavailable).
Discussion
Case Discussion
Peripheral polyneuropathy is a rarely documented complication of CVID. A number of alternative explanations were considered. Given the patient’s immunocompromised state, infectious causes of neuropathy were considered, although the patient’s history, exam, and workup did not suggest this. Further workup also ruled out nutritional deficiencies such as vitamin B12. Various medications such as digoxin, phenytoin, statins, cisplatin, vincristine, isoniazid, and amiodarone can cause neuropathy, which is not present in our patient (Azhary et al., 2010). An inherited neurologic disorder such as metachromatic leukodystrophy, Charcot–Marie–Tooth, or other hereditary motor sensory neuropathy variants can cause neuropathy but were unlikely given both patient presentation and negative family history (Azhary et al., 2010). In addition, the patient’s history did not support hereditary neuropathy with liability to pressure palsies. Diabetes is a widely known cause of neuropathy, but she had no signs or symptoms of diabetes and HbA1c was normal (Pinzur, 2011). While nerve root compression is a potential source of symptoms, her history, physical exam, and lack of correlating imaging findings did not support this. Finally, peripheral neuropathy commonly affects older adults. It is estimated that 28% of adults 70 to 79 years old and 35% of adults ≥80 years have neuropathy; our patient was 62 years old at the onset of symptoms (Gregg et al., 2004). Although age can be a risk factor for development of neuropathy, it is more commonly symptomatic in those with concurrent disease such as diabetes; in fact, lower extremity disease is twice as common in concurrent diabetes (Gregg et al., 2004; Popescu et al., 2016), which is absent in our patient. Having ruled out other explanations, we concluded this patient’s peripheral neuropathy was related to CVID.
Review of Literature
Neurological complications can occur in primary and combined immunodeficiency diseases (Al-Herz et al., 2011; Hermaszewski & Webster, 1993). There is a paucity in the literature of reported cases, and most are CNS manifestations (Kondo et al., 2005; Rudge et al., 1996). The underlying pathophysiology of peripheral nerve disease in CVID is not well understood but several proposed mechanisms exist (Knight & Cunningham-Rundles, 2006). It is thought that peripheral neuropathy may be caused by both a cellular and humoral reaction to peripheral nerves (Özdemir, Okan, & Kilic, 2012). There is a well-documented connection between CVID and autoimmune disease. Some CVID patients produce a limited amount of self-reactive antibodies causing various autoimmune diseases such as autoimmune hemolytic anemia and sarcoid-like granulomatous disease (Knight & Cunningham-Rundles, 2006). In a similar fashion, it is possible in our case that the patient may have produced autoantibodies against neural components resulting in peripheral neuropathy. Enteroviral infection is also proposed (Rudge et al., 1996), but this case was in the setting of meningoencephalitis (neither enterovirus infection nor CNS disease was present in our case). Finally, peripheral neuropathy in CVID could be secondary to a complex response to IVIg treatment (Rudge et al., 1996). Although neurologic complications have been noted in a number of patients receiving IVIg, peripheral neuropathy has not been documented (Ziegner et al., 2002). IVIg may cause neurologic symptoms that progress into neurodegenerative disorders; however interestingly, high-dose IVIg actually improved peripheral neuropathy symptoms in a previous case of CVID (in pediatric chronic inflammatory demyelinating polyneuropathy [CIDP]; Özdemir et al., 2012; Ziegner et al., 2002). Due to the complexity of this interaction, the interplay between IVIg, CVID, and peripheral neuropathy is unknown.
There is one case of adult axonal peripheral neuropathy due to CVID (Larner et al., 2000). Larner and colleagues reported a case of peripheral neuropathy in a 79-year-old man diagnosed with CVID 25 years previously. The patient had a similar sensorimotor polyneuropathy to our case (mainly axonal). Differential diagnosis in this case included chronic progressive myelitis and ganglionopathy before the authors concluded the patient’s peripheral neuropathy secondary to CVID. Their explanation proposed a connection between the patient’s symptoms and an unknown autoimmune mechanism.
One case of CIDP in CVID is reported; however, this was in a pediatric patient aged 7 years (Özdemir et al., 2012). Although the pathophysiology is certainly of interest as the authors proposed a similar autoimmune response, childhood CIDP does not as easily apply to adult CVID and its complications.
Conclusion
To our knowledge, this is the second known case of peripheral neuropathy as a complication of CVID. It also illustrates the often vague and nonspecific nature of initial neuropathy symptoms. Lacking a detailed history, physical, and focused diagnostic workup, the diagnosis in our patient remained unclear for several years and she underwent a nonspecific and costly workup without an answer. Impaired peripheral nerve function from neuropathy is known to result in poor physical performance, impaired balance, and increased fall risk, while also significantly affecting independence and activities of daily living in older patients (Lauretani et al., 2006; Popescu et al., 2016; Resnick et al., 2002; Strotmeyer et al., 2006). Thus, providers need to be aware of the potential peripheral nervous system complications of primary immune deficiencies such as CVID and must be prepared to initiate a focused workup. Awareness of this possibility will lead to a more prompt workup and earlier diagnosis, and also positively impact quality of life and daily activities in CVID patients.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.