
Abstract
Localized amyloidosis is a rare benign disease characterized by extracellular deposition of misfolded proteins in a specific organ without systemic involvement, which might lead to organ dysfunction. Patients with localized amyloidosis do not develop systemic diseases such as cardiac, renal, hepatic, or nerve involvement. Typically, the localized disease is managed by surgical resection, while the systemic disease is treated with a combination chemotherapy and immunotherapy. However, some patients can have challenging clinical presentations, delaying diagnosis and organ dysfunction, and they might require systemic therapeutic intervention. There is a significant paucity of knowledge and data regarding clinical manifestations and the course of localized amyloidosis in various organs, and management can also be challenging. It is crucial to appropriately balance effective therapy with patient safety as treatment can lead to toxicity. Here, we discuss a series of patients who were presented with localized amyloidosis and one of whom needed systemic therapy.
Introduction
Amyloidosis is a rare disease characterized by extracellular deposition of misfolded proteins leading to organ dysfunction. 1 The most common causes of amyloidosis are AL (light chain) and AA (amyloid A protein) amyloidosis. AL represents 78% of new amyloidosis cases in the United States per year, while AA amyloid makes up 6% of cases. 2 AA is reactive amyloidosis due to chronic inflammatory diseases such as rheumatoid arthritis (RA). AL amyloidosis is caused by misfolded light chains depositing in diffuse organs, causing dysfunction. 2
Amyloidosis describes a rare condition in which local amyloid deposits in tissues such as lungs, urogenital systems, or skin; these deposits are derived from monoclonal light chains, but there is no evidence of an underlying systemic clonal plasma cell disorder (Figure 1). 3 Amyloidosis can be either localized or systemic. Patients with localized amyloidosis do not develop systemic manifestations such as the deposition of amyloid in other organs outside the primary site such as cardiac, renal, hepatic, or nerve involvement, and treatment is primarily surgical excision. 4 Due to their rarity, much of our understanding of amyloidosis is limited to case reports and series. A biopsy remains the gold standard for diagnosis, with characteristic findings of Congo red-positive amyloid deposits that exhibit apple-green birefringence under polarized light. 3

Pathobiology of amyloidosis. Normally folded proteins undergo misfolding and aggregate into β-pleated sheet-rich amyloid fibrils, which accumulate extracellularly. These fibrils deposit in tissues, disrupt cellular architecture, and contribute to organ dysfunction. Created in BioRender. Dermarderosian (2025), https://BioRender.com/6c67lrb.
Systemic amyloidosis has the potential to affect almost any organ system, though cardiac involvement is the leading cause of morbidity and mortality. 5 Patients with AL amyloidosis are more symptomatic. 6 Unlike other hematological malignancies, notably multiple myeloma, morbidity and mortality in amyloidosis are not caused by plasma cell proliferation but, rather, the toxic effects of monoclonal protein deposition, which leads to end organ damage, with cardiac function being the most important determinant of survival. 7
Amyloidosis is a difficult diagnosis to make as patients often present asymptomatic or symptoms are insidious and nonspecific. Fatigue, light-headedness, and weight loss are just a few common nonspecific symptoms, with others being less common, such as jaw claudication, peripheral neuropathy, carpal tunnel syndrome, and alopecia. 8 Macroglossia and periorbital purpura occur in about one-third of cases but are virtually pathognomonic for AL amyloidosis. 6 Upon diagnostic workup, histology demonstrates green birefringence under cross-polarized light on Congo red staining of the tissue biopsy; however, laser capture mass spectroscopy is now the gold standard for diagnosis. 8
Typically, localized disease is managed by surgical resection, while systemic disease is treated with combination chemotherapy and immunotherapy. However, some patients can have challenging clinical presentations. Here, we discuss a series of patients who were presented with symptomatic localized amyloidosis (Table 1).
Patient Summary.
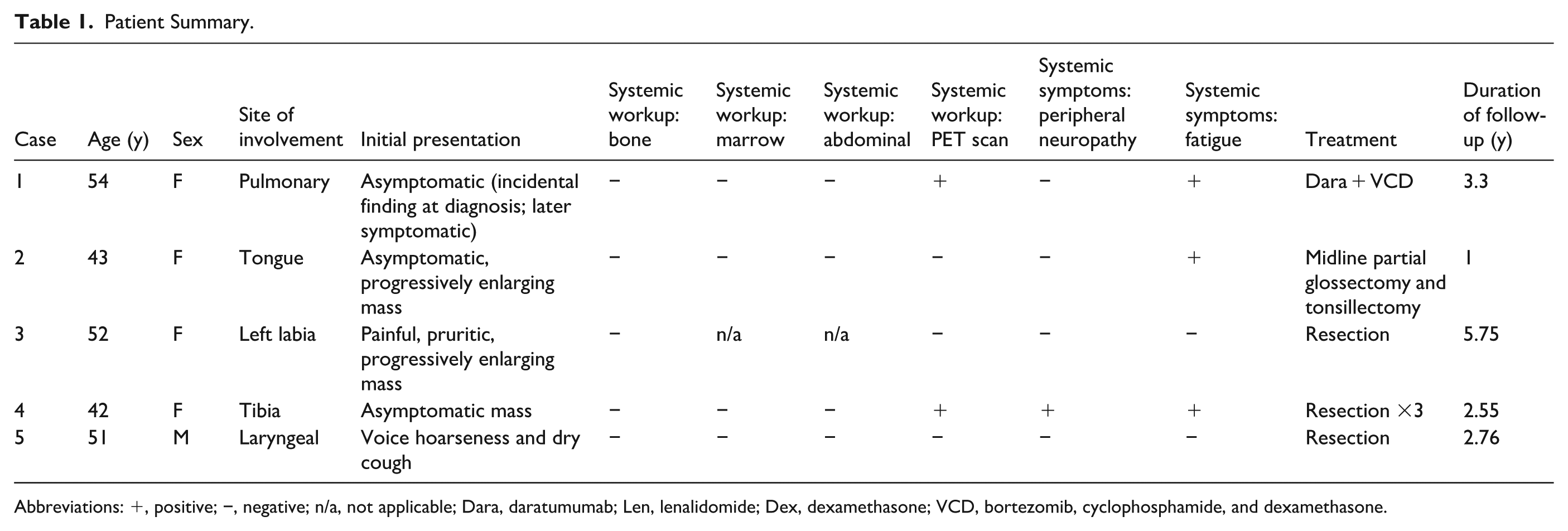
Abbreviations: +, positive; −, negative; n/a, not applicable; Dara, daratumumab; Len, lenalidomide; Dex, dexamethasone; VCD, bortezomib, cyclophosphamide, and dexamethasone.
Case Presentation
Case Number 1
A 54-year-old Hispanic female with Sjogren’s and arthralgias presented for evaluation of pulmonary nodules. In early 2022, she was incidentally found to have bilateral 1 to 2 cm pulmonary nodules on a computed tomography (CT) chest. Her only symptoms were fatigue at the time which was not interfering with activities of daily life.
A core biopsy of the right lung confirmed amyloidosis (Figure 2). Positron emission tomography (PET) scan showed a mildly FDG (F-18 fluorodeoxyglucose) avid 1.4-cm right lower lobe nodule and additional nodules with minimal FDG activity in the right middle and upper lobes, left upper lobe, and lingula. An abdominal fat biopsy was negative for amyloidosis, indicating localized disease. A bone marrow (BM) aspiration and biopsy showed polyclonal plasma cells. With histological confirmation and ruling out systemic disease, she was diagnosed with localized amyloidosis. Over the next few years, she was followed up very closely with regular appointments and blood work for 2 years until she presented with worsening shortness of breath and chest pain. Due to the increasing symptoms of her localized pulmonary amyloidosis, including worsening dyspnea that was affecting her activities of daily life, she was started on a combination of immunotherapy and chemotherapy regimens commonly used for systemic amyloidosis: daratumumab, velcade, cyclophosphamide, and dexamethasone (Dara-VCD) for 6 cycles. She tolerated the treatment despite some side effects, including peripheral neuropathy and dental pain which resolved with conservative measures.

Histopathologic findings from a right lung mass core biopsy demonstrating features consistent with amyloidosis. (Left) Hematoxylin and eosin stain shows amorphous eosinophilic extracellular deposits. (Right) Congo red stain highlights the deposits with red-orange staining under non-polarized light, supporting the diagnosis of amyloidosis.
In 2024, she was presented with worsening shortness of breath and chest pain. Due to the increasing symptoms of her localized pulmonary amyloidosis, she started a combination of immunotherapy and chemotherapy regimens commonly used for systemic amyloidosis: Dara-VCD. She tolerated the treatment despite some side effects, including peripheral neuropathy and weakness in her teeth. Nodules decreased in size, and symptoms resolved. She completed 6 cycles and received a CT scan following completion of therapy.
She continued with her current treatment regimen of Dara-VCD. The patient started gabapentin to manage worsening peripheral neuropathy resulting from her chemotherapy. Complete Blood Count with Differential (CBC) with differential, Comprehensive Metabolic Panel (CMP), Lactate Dehydrogenase (LDH), and uric acid are done every 4 weeks. A repeat PET scan conducted 7 months after initiating treatment demonstrated a response to treatment, with a decrease in FDG activity in the right lower lobe nodule, which is now measuring 2.3 cm. The additional nodules demonstrate no significant FDG activity, though the smaller nodules may be below PET resolution. The plan remains to complete 2 years with daratumumab and hyaluronidase for 2 years, followed by maintenance with Pomalidomide.
Case Number 2
A 43-year-old Hispanic female with a history of anemia and bilateral osteoarthritis presented with an increasing posterior mass on her tongue. The patient first noticed a mass on the middle posterior tongue when drinking water 3 months before presenting to the clinic. The mass started as a dot and has been slowly increasing in size. Initial examination of the oral cavity by ENT showed a 1-cm firm, nodular-type mass of the midline posterior one-third of the tongue. She denied pain, changes in taste, and trauma to the area. A review of systems was positive for systemic symptoms, including weakness and headache. However, those symptoms were related to iron deficiency anemia secondary to heavy periods.
A biopsy of the midline posterior tongue lesion was positive for amyloid (Figure 3). BM biopsy was negative for amyloid and plasma cell dyscrasias. Abdominal fat biopsy was negative for amyloid. PET-CT scan showed increased FDG activity of the mildly prominent palatine tonsils as well as along the mucosal surfaces of the oral cavity, of uncertain significance and possibly physiologic, and no obvious FDG abnormality within the tongue below the mucosa. Her complete blood count revealed iron deficiency anemia, likely secondary to menorrhagia.

Histologic section of tongue tissue from medial glossectomy demonstrating subepithelial nodular deposits of amyloid consistent with glottic amyloidosis. The amyloid appears as amorphous eosinophilic material beneath the epithelium (hematoxylin and eosin stain, 10× magnification).
Given the negative BM and abdominal fat pad biopsies, this patient was diagnosed with localized amyloidosis. She underwent resection via midline partial glossectomy and tonsillectomy with no complications. The patient is doing well, and subsequent lab studies and imaging have not shown any evidence of disease.
Case Number 3
A 52-year-old Hispanic female with a history of diabetes mellitus, anemia, hysterectomy and oophorectomy, and hypertension presented to the hematology–oncology clinic for newly diagnosed vulvar amyloidosis after excision. The patient first noticed a small mass on her left labia that had been increasing in size. She endorsed pain and pruritus at the site. An initial biopsy done by outside dermatology showed vulvar amyloidosis. Later, an excisional biopsy was done by gynecology, and a 5.5 × 2 × 0.5 cm mass was resected. The margins were not noted in the report. The review of systems was negative for systemic symptoms, including peripheral neuropathy. The physical exam post-resection was unremarkable, with no mass detected on the pelvic exam.
A biopsy of the left labial mass was positive for amyloid (Figure 4). BM aspirate/biopsy was negative for amyloidosis, including no evidence of plasma cell dyscrasia or amyloid deposition. The immunophenotyping flow cytometry and immunohistochemistry showed no monotypic plasma cells. The vulvar lesion was resected without complications. The patient is to repeat labs every 6 months, including CBC with diff, CMP, LD, uric acid, SPEP, immunofixation, and serum light chain panel. Follow-up in the hematology clinic in 12 months. However, the patient has been lost to follow-up.

Histologic section from a punch biopsy of the left labia minora demonstrating findings consistent with dermatophytosis and underlying nodular amyloidosis. The overlying stratum corneum contains fungal hyphae (highlighted on PAS, not shown), confirming concomitant dermatophytosis. Congo-red staining of the deposits was congophilic with weak apple-green birefringence, and immunostains revealed a kappa predominant plasma-cell infiltrate, supporting light-chain-related amyloid deposition.
Case Number 4
A 42-year-old Hispanic female with morbid obesity, GERD, anxiety, depression, migraine, and Raynaud syndrome presented for evaluation of tibial amyloidosis and to rule out systemic amyloidosis.
The patient was found to have a soft tissue mass on her anterior tibia on a physical exam by the primary care physician. MRI was ordered and revealed a 3.5 × 2.9 × 2.0 cm soft tissue lesion. In June 2022, the mass was biopsied and resected. Pathology at the time showed amorphous eosinophilic material consisting of amyloidosis. The lesion abutted the deep and superficial margins. She was hospitalized a month later due to cellulitis at her surgical site, which resolved after incision and drainage, and antibiotics. Later that year, the patient reported 2 new masses on her right lower extremity, which were increasing in size. A repeat MRI of the tibia/fibula revealed 3 separate lesions, a 2-cm nodule laterally, and a 5-cm thickening of soft tissue more posterolateral. In November 2022, 2 soft tissue masses, measuring 3 × 6 and 2 × 2 cm, respectively, and a recurrent pre-tibial amyloidosis soft tissue mass, measuring 3 × 7 cm, were resected. She continued to endorse pain in the right lower extremity post-resection. In November 2023, the patient underwent resection for new masses in the lower right extremity. She had a repeat biopsy done in March 2024.
She most recently presented with increased forgetfulness, nonspecific for amyloid, but upon asking a review of systems questions, she was positive for night sweats, low-rated fever, fatigue, and neuropathy in the right leg. The biopsy of the initially resected right medial tibial mass revealed amorphous eosinophilic material, consistent with AL (lamba)-type amyloidosis (Figure 5). Congo red-positive amyloid deposits were present. BM aspirate biopsy and abdominal fat pad biopsy were negative. A PET scan revealed mild FDG activity related to anterior right lower extremity biopsy, diffuse FDG activity within the spine and pelvis of uncertain significance and mildly enlarged FDG-avid bilateral level II cervical lymph nodes, which may be reactive. The antinuclear antibody (ANA) panel was positive for ANA and anti-centromere antibodies.

Histologic sections of a right tibial mass demonstrating nodular deposits of amorphous eosinophilic material consistent with amyloid. (Left) Hematoxylin and eosin stain shows pink, amorphous extracellular deposits with surrounding moderate histiocytic reaction. (Right) Sulfated Alcian blue stain highlights the amyloid deposits in turquoise, confirming the presence of amyloid. No viable malignancy was identified.
As mentioned above, the patient underwent resection 3 times.
The patient had a biopsy in March 2024. The patient is being followed up by an outside hospital.
Case Number 5
A 51-year-old male with a past medical history of frontal bossing and hearing loss presented to the hematology oncology clinic for evaluation of amyloidosis.
He first presented to his otolaryngologist for worsening hoarseness of his voice and a dry cough that had been ongoing for over 8 months. He had a biopsy of the right infraglottic mass which confirmed amyloidosis. At the time of his initial consultation, he denied any symptoms of neuropathy and he was very active, mobile, and able to complete activities of daily life without difficulty. The right infraglottic mass biopsy showed submucosal deposits of homogenous eosinophilic material suggestive of amyloidosis. It was also positive on Congo red stain, showing apple green birefringence upon polarization. A BM aspiration and biopsy was performed to evaluate for the presence of clonal plasma cells in the BM and amyloid deposition, which showed no evidence of amyloidosis or other malignancies. The marrow was hypocellular for his age with active trilineage hematopoiesis with 3% polytypic plasma cells. He also underwent abdominal fat pad biopsy to determine the amyloid subtype, which was negative for systemic amyloidosis.
Patient proceeded to have surgical resection of his localized amyloidosis. His surgery was completed successfully without complications, and he did not need chemotherapy after.
The patient is doing well and continues to follow hematology oncology for surveillance with labs every 6 months. We continue to monitor his complete blood count, metabolic panel, lactate, uric acid, myeloma panel, and beta-2 microglobulin.
Discussion
The 5 cases we presented demonstrate how localized amyloidosis can present and how management can differ based on the patient’s presentation and localization of the disease. While some are monitored, others are candidates for resections and some for medical management with systemic chemotherapy like Dara-VCD.
Systemic amyloidosis is a rare protein misfolding and deposition in organs, causing dysfunction. There are over 15 types of amyloidosis that can affect various organs, including the heart, kidney, liver, gastrointestinal tract, lungs, skin, and soft tissue. Amyloidosis has been a difficult diagnosis to make in the past due to its insidious onset and nonspecific findings. However, with new mass spectrometry-based shotgun diagnostics, we can better diagnose the treatment of the disease. 9
Many studies have sought to address the difficulty in considering amyloidosis as a differential for many nonspecific symptoms and incidental findings, leading to failure to recognize and treat the disease earlier in its course. 10 Furthermore, management with limited data from clinical trials poses another challenge, making this an important topic for discussion. Diagnosis for systemic disease requires histological confirmation of Congo red deposits, typically from fat aspiration and/or BM biopsy. Organ biopsies can diagnose localized diseases. Muchtar et al looked at 612 patients with AL amyloidosis to characterize the tissues used to establish a diagnosis. They found that Congo-red sensitivity was highest for tissue biopsies at 70% to 100%; however, fat aspiration and BM biopsy combined was 87%. Per their analysis, fat aspiration was underutilized for histological confirmation of amyloidosis, and the high rate of organ biopsies represents a failure to recognize the disease. 10 On the other hand, our case series highlights the importance of tissue biopsy in confirming the diagnosis, especially in localized disease. Fat biopsy and BM biopsies were unremarkable in our patient set; however, they each presented with symptomatic disease, making their tissue biopsies a vital component of their positive outcomes.
For pulmonary amyloidosis, AL is most common, and the nodular subtype responds well to excision, while the diffuse alveolar-septal and tracheobronchial amyloidosis requires a system approach. 11 In Case 1, localized amyloidosis was treated with systemic chemotherapy, an unconventional approach in such cases. This decision was made because the patient was experiencing significant symptoms of chest pain and dyspnea despite only having a localized disease. Furthermore, the pulmonary nodules were unresectable. Dara-VCD reduces amyloid production, engages the immune system (daratumumab), and induces cell death (bortezomib and cyclophosphamide). Adding dexamethasone improved the quality of life during treatment by making treatment more tolerable. The total duration of treatment was planned for 2 years with the aim to achieve deep and sustained hematologic response given the extent of the patient’s symptomatic disease. Pulmonary amyloidosis is a spectrum, but the current literature treats localized disease with surveillance or localized treatment. However, there exists a handful of patients presented in case series, such as Khoor et al, particularly that of a 62-year-old male with multiple pulmonary nodules in the setting of lymphoplasmacytic lymphoma treated with systemic chemotherapy. These lesions were found to be amyloid-like light chain deposits associated with an underlying clonal lymphoproliferative disorder. 12 This is a case that is mentioned to highlight the importance of distinguishing nodular amyloidosis from amyloid light chain deposits.
This case is of further interest because pulmonary nodules are typical indolent event with multifocal lesions. Typical management involves observation, periodic imaging, bronchoscopy with debulking, or localized therapies such as radiation. Our consideration of systemic therapy is, therefore, not intended to redefine standard practice, but to highlight select scenarios in which persistent or progressive local disease may justify intervention beyond surveillance alone.
In Case 2, we discussed a patient with amyloidosis of the tongue. It is more likely that the patient has systemic amyloidosis due to the rarity of localized amyloidosis in the tongue. 13 A case series in 2013 only found 12 cases of such literature. 13 Systemic amyloidosis with tongue involvement is more typical. 13 Literature does suggest that long-term dialysis can contribute to oral amyloidosis of the tongue as well. 14 However, if she is confirmed to be negative for systemic infiltration, observation or resection is the mainstay of treatment. 13 Although a simple surgical resection is the choice of treatment for localized amyloidosis of the tongue, this procedure may be difficult in this patient due to the size of the mass. 15 If resection is not possible due to multifocal disease or difficult anatomy, an alternative treatment option must be considered such as systemic chemotherapy, as seen in Case 1. However, the patient from Case 2 responded well to surgical resection and did not require further intervention. This case of localized amyloidosis with involvement of the tongue is particularly interesting due to its rarity, given that there was no systemic involvement.
Localized amyloidosis, as seen in the patient discussed in Case 3, is characterized by amyloid deposits confined to a single organ or tissue without evidence of systemic involvement. This was confirmed by a negative BM biopsy, absence of plasma cell dyscrasia, and negative systemic workup, including serum and urine protein electrophoresis and immunofixation studies. The localized form of amyloidosis is thought to arise from localized plasma cell proliferation and subsequent amyloid deposition, although the exact pathophysiology remains poorly understood.
Localized amyloidosis of the vulva has previously been largely discussed only in case reports and series. Quddus et al found that keratin-associated amyloid materials (CK5 and CK14) were unique to localized vulvar amyloidosis. 16 A prior hypothesis is that these unique amyloid deposits are derived from tumor cells. These specialized keratin-derived cells have been seen in squamous cell cancer, endocrine tumors, Hodgkin lymphoma, and renal cell carcinoma. 16
Treatment options for localized vulvar amyloidosis are not well established due to the rarity of the condition. In this case, surgical excision of the lesion was performed successfully without complications. Surgical excision is often considered the treatment of choice for localized lesions, particularly when symptoms such as pain or pruritus are significant. However, recurrence remains a concern, as localized amyloidosis has been reported to recur even after complete excision.
Long-term follow-up is essential in cases of localized amyloidosis to monitor for recurrence or progression to systemic disease. Our patient was counseled to obtain periodic laboratory assessments (CBC, CMP, LDH, serum light chains, and SPEP); however, she was ultimately lost to follow-up. This significantly limits our ability to comment on long-term outcomes or relapse risk and underscores a common real-world challenge in managing rare diseases. Consequently, our conclusions are descriptive and hypothesis-generating rather than definitive. Future studies should explore strategies to identify and retain patients at high risk of being lost to follow-up to improve outcome assessment.
In conclusion, Case 3 highlights the rarity of vulvar amyloidosis and the importance of early recognition, histopathological confirmation, and appropriate management. Regular follow-up remains essential to monitor for recurrence and systemic involvement.
Though there are limited data on both systemic and localized treatment and management of amyloidosis, various trials have been conducted to assess the hematological response to therapy. Kastritis et al conducted a phase III trial where 388 patients with newly diagnosed systemic AL amyloidosis underwent randomization with either 6 cycles of bortezomib, cyclophosphamide, and dexamethasone either alone or with subcutaneous daratumumab for 6 cycles followed by single agent daratumumab. Their results found that the addition of daratumumab was associated with higher frequencies of hematologic complete response, defined as involved serum free light chain levels less than the upper limit of normal with negative immunofixation by serum and urine (53.3% vs 18.1%, relative risk ratio 2.9, 95% CI 2.1-4.1, P < .001) and survival free from major organ failure or progression. 17 The standard of care for AL amyloidosis has typically consisted of cyclophosphamide, bortezomib, and dexamethasone, but now, novel anti-proteasome inhibitors, immunomodulators, monoclonal antibodies, and small molecule inhibitors are emerging as supplements to the current standard of care. 18 The goal of systemic treatment is to achieve very good partial light chain responses.
The challenge remains that diagnosis is often delayed despite having effective therapeutic strategies. This results in organ failure before initiation of treatment. However, trials are in process to find antibodies that can deplete the deposited fibrils. 19
Case 4 narrates a patient whose amyloidosis was diagnosed as a soft tissue amyloidoma of the right lower leg. Amyloidoma, an amyloid tumor, is the rarest form of amyloidosis, limited only to case reports, and its presentation in the extremities is also rare. 20 Additionally, some amyloidoma are associated with plasmacytomas. 21 This patient’s treatment of resections is consistent with what has been seen in literature as the treatment for localized amyloidosis. 20 The patient required multiple resections for recurrent tibial masses, which raises the question, could they have benefited from systemic treatment. In this case, each resection was amenable to complete resection without impacting that patient’s functional status or organ tissue compromise. This case provides an alternative management strategy to that of Case 1 and proposes that locally aggressive disease can be managed with local therapy alone. Together, the 2 cases underscore the need for patient-centered decision making and balancing recurrence risk, symptom severity, and clonal evaluation.
Amyloidoma, as mentioned previously, is rare. Clinically, they can present as a solitary mass, often be mistaken for other neoplastic processes such as chondrosarcomas. 22 Pathologically, the tissue biopsy showed amorphous eosinophilic material that exhibits apple-green birefringence under polarized light on Congo red staining, consistent with amyloid deposits. 23 Prognosis is favorable following surgical excision and in the absence of systemic disease but, again, there is little known about the impact systemic therapy may have on these cases. These amyloidomas can cause progressive complications through mass effect or obstruction when found at critical anatomical sites. Resection can be complex in these special circumstances, and systemic therapy may be considered not to prevent systemic progression, but to reduce ongoing local light chain production that contributes to recurrent deposition and symptom burden. Thus, our reference to systemic therapy is not a departure from the amyloidoma paradigm, but a recognition that select patients with function-threatening disease may warrant broader plasma cell-directed treatment beyond local intervention.
Finally, Case 5 presented a classic case of localized amyloidosis found in an infraglottic mass. In an analysis on localized amyloidosis that evaluated 5551 patients, the most commonly involved sites were urothelial and laryngeal tissue (21% and 14%, respectively). In this study, 61% of patients were successfully treated with resection, similar to our patient presented in Case 6, while 21% were monitored for disease progression or worsening symptoms. Ten-year survival was 78% and no patients had progression to systemic disease, though some did have progression of localized disease. 4 The challenge with localized disease is the possibility to recurrence. Seventeen percent of patients in this study recurred at a median duration of 41 months after diagnosis with 63% recurring within the first 5 years. Furthermore, in this large series, laryngeal amyloidosis was more likely to have recurrence than other organ involvement. 4 This supports why the patient in Case 6 continues to be evaluated in 6-month intervals. There remains paucity of knowledge on how to approach recurrent localized disease, but newer approaches with chemotherapy are providing alternatives in treatment for these patients. 17
The patient is suspected to have an underlying autoimmune disease, as evidenced by her positive ANA panel, and is being treated with Plaquenil. Although amyloids are typically thought to be harmful proteins in the context of autoimmune diseases due to their association with the pro-inflammatory state, research has shown that certain amyloids may be protective and induce an anti-inflammatory state as well. 24 These observations, however, remain largely theoretical and have not changed current clinical practices surrounding the management of localized amyloidosis. Addressing the pathological protein deposition with surgery remains the mainstay treatment. Thus, if considering systemic therapy in select cases, the autoimmune management must be carefully considered to avoid exacerbating either condition.
Reports have recognized the association of autoimmune diseases and amyloidosis. Primary Sjorgren’s disease, in particular, is associated with more than just B-cell lymphoma. High serologies for autoimmune disease should also prompt a workup for localized amyloidosis. 25 Cases 1 and 5 both reported patients with rheumatological conditions concurrently with amyloidosis. AA amyloidosis is known to be a complication of chronic inflammatory conditions, such as RA, systemic lupus erythematosus, and even inflammatory bowel disease. This is because serum amyloid A is an acute-phase reactant that eventually may deposit as amyloid. 26 AL amyloidosis occurs more in the context of Sjogren’s disease as a result of deposition on immunoglobulin light chains produced by clonal plasma cells. 27 In conclusion, amyloidosis suspicion should increase in patients with a known history of rheumatological conditions.
Overall, there are some limitations that should be addressed prior to using this case series to draw conclusions from. The limited duration of follow-up for a majority of these patients redistricts our ability to assess the long-term benefit of Dara-VCD and surgical resection. It also makes it difficult to fully characterize the natural history of localized amyloidosis. However, we can say that initial resolution and stabilization of symptoms was observed in all patients. As a result, the findings mentioned in this study should be regarded as descriptive and preliminary in nature. Future collaborative or registry-based studies with standardized, long-term follow-up protocols are needed to clarify true disease trajectories and sustained treatment outcomes.
Prior literature, including the series by Kourelis et al, supports observation or minimally invasive intervention as a reasonable default approach. However, our case illustrates a scenario in which more aggressive systemic therapy was considered due to extensive local involvement and organ-specific morbidity. Although localized amyloidosis is typically indolent, emerging reports suggest that a minority of patients may experience persistent or progressive symptom burden despite conservative measures. Here, systemic therapy may be employed to suppress ongoing light chain production and mitigate further amyloid deposition. We acknowledge, however, that without long-term follow-up, we cannot determine whether this approach confers durable benefit in our patient. This highlights the need for prospective comparisons between observation, local treatments, and systemic therapy in higher-risk presentations of localized amyloidosis.
Conclusion
This case series provides valuable insight into the diverse presentation and management of amyloidosis across a varied patient population. While some patients exhibited a remarkable response to systemic therapy, particularly those who were symptomatic at presentation, others benefited from surgical resection. Notably, the use of Dara-VCD, typically reserved for systemic amyloidosis, demonstrated promising outcomes in cases of biopsy-proven localized disease, suggesting its potential as a treatment option in select cases. This raises the question of whether systemic therapy might be considered in highly selected patients with symptomatic, unresectable localized amyloidosis, though further study is needed to define appropriate patient selection criteria and to establish efficacy and safety. These findings contribute to a broader understanding of therapeutic approaches for localized amyloidosis.
Footnotes
Acknowledgements
We appreciate the staff at the Loma Linda University Cancer Center and Department of Pathology.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent to Participate
Written informed consent was obtained from all patients for the inclusion of their de-identified clinical data in this case series and for publication in scientific literature.
Consent for Publication
Verbal informed consent was obtained from the patients for their anonymized information to be published in this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.