
Abstract
22q11.2 deletion syndrome is a multifaceted disorder most characterized by congenital cardiac anomalies, immunodeficiency, and psychiatric conditions. Endocrine abnormalities such as hypoparathyroidism and growth hormone deficiency are well documented, but hypogonadism remains rarely reported in this patient population. Only 1 case of hypogonadism has been reported in a patient with multiple other comorbidities which may have contributed to the condition. The relationship between 22q11.2 deletion syndrome and hypogonadism is not well understood. We report 2 male patients with 22q11.2 deletion syndrome and low testosterone levels. The first patient was a 25-year-old male with Tetralogy of Fallot and hypoparathyroidism who presented with balanitis and was found to have low testosterone. Evaluation for causes, including pituitary imaging and hormone panels, was unremarkable. The second patient was a 20-year-old male with a history of growth hormone deficiency and hypogonadism, scoliosis, and neurodevelopmental disorders who had low testosterone levels. No identifiable causes were found. Mechanisms include disruptions in the hypothalamic–pituitary–gonadal axis during embryonic development or testicular dysfunction. Impaired function of synaptosomal-associated protein 29, a gene located within the 22q11.2 region, may contribute to testosterone deficiency. The rarity of reported hypogonadism in 22q11.2 deletion syndrome suggests that it may be underdiagnosed due to a lack of routine screening protocols. Further studies evaluating testosterone, LH, and FSH levels in this population are warranted to establish prevalence and determine whether routine endocrine assessment should be incorporated into clinical guidelines.
Introduction and Background
22q11.2 deletion syndrome is a multifaceted genetic disorder with an estimated prevalence of 1 in 3000 to 1 in 6000 live births, making it one of the most common microdeletion syndromes. 1 The syndrome consists of a deletion in the 22q11.2 region, most often manifesting as a 3 Mb hemizygous deletion referred to as the typically deleted region. 2 Genetic variants in multiple genes, such as TBX1 and DGCR8, arise as a result of a deletion in this region, leading to heterogeneous clinical presentations. 1 Common presenting symptoms include cardiac defects, palatal abnormalities, learning difficulties, immunological deficiencies, psychiatric disorders, and learning disabilities.3,4 Fetal loss and infant death have also been reported. 5
Reproductive and endocrine abnormalities in the 22q11.2 deletion syndrome may include inguinal hernias, hypoparathyroidism, thyroid abnormalities, growth hormone deficiency, intrauterine growth restriction, and short stature. 1 Absent uterus has been reported in females with 22q11.2 deletion syndrome. 1 In males with 22q11.2 deletion syndrome, cryptorchidism, hypospadias, 1 azoospermia, and micropenis have been reported. 5 Males with 22q11.2 deletion syndrome have a decreased reproductive fitness. However, those without schizophrenia or intellectual disability maintain reproductive fitness comparable to the general population. 1 Impaired male fertility in these patients may be due to the absence of synaptosomal-associated protein 29 (SNAP29), a protein that plays an important role in the testis/body ratio and spermatogenesis. 5
Hypogonadism is very rare in patients with the 22q11.2 deletion syndrome as there has been only 1 reported case of a 35-year-old male with late-onset hypogonadism. 5 This patient was diagnosed with a pituitary adenoma 1 year after presenting with hypogonadism. He then experienced decreased testosterone levels a year later while being treated with bromocriptine. The patient also had a history of type 2 diabetes mellitus, obesity, and chronic kidney disease, all factors that may cause hypogonadism. 5 The only reported case of hypogonadism in patients with 22q11.2 deletion syndrome can be explained by many different factors which may have led to the hypogonadism that was seen in this patient.
Case Presentation #1
Patient is a 25-year-old male with history of 22q11.2 deletion syndrome with congenital cardiac diagnosis of Tetralogy of Fallot as well as anomalous left pulmonary artery (LPA) from the ascending aorta. Patient has a past surgical history of complete repair of Tetralogy of Fallot and anomalous LPA from the ascending aorta (surgical details not available), right ventricular outflow tract reconstruction with an 18 mm valved pulmonary homograft, left pulmonary artery plasty with homograft, resection of right ventricular muscle bundles and closure of patent foramen ovale, and a right heart catheterization with balloon angioplasty of LPA. During the period of evaluation, the patient’s medications included metoprolol extended release 25 mg daily for treatment of congenital heart disease and calcitriol 25 µg BID for treatment of hypocalcemia secondary to hypoparathyroidism. Previously, patient was treated with GH during high school for short stature. Patient has no pertinent family history.
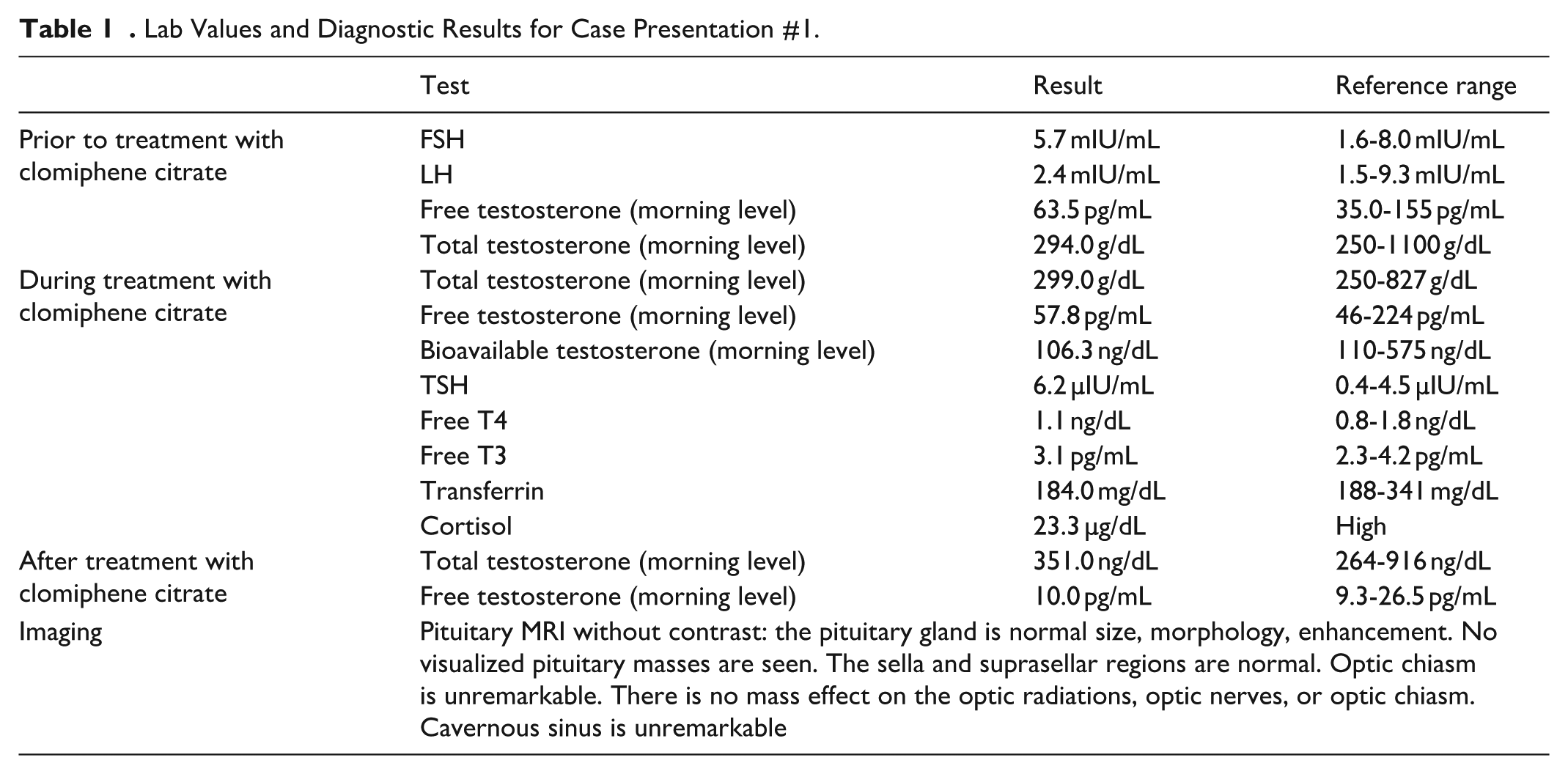
Patient initially presented with recently discovered hypospadias with severe balanitis and phimosis. This condition was ultimately treated surgically with circumcision. After the surgery, the patient presented for evaluation of hypoparathyroidism. Laboratory tests were ordered, which revealed relatively low testosterone levels (294 ng/dL) alongside normal FSH (5.7 mIU/mL) and LH (2.4 mIU/mL). Despite normal FSH and LH levels, a workup was ordered to investigate potential causes of hypogonadism, including an iron panel to assess for hemochromatosis, prolactin levels to evaluate for hyperprolactinemia, cortisol levels to screen for adrenal insufficiency, and a pituitary MRI to rule out structural abnormalities such as pituitary adenomas. All additional testing did not reveal any causes for hypogonadism. Patient was treated with clomiphene citrate 50 mg daily for low testosterone levels. Testosterone therapy was also discussed with patient, but it is unclear at this time whether he decided to pursue that therapy (Table 1).
Lab Values and Diagnostic Results for Case Presentation #1.
Case Presentation #2
Patient is a 20-year-old male with a history of 22q11.2 deletion syndrome with growth hormone deficiency and hypogonadism, scoliosis, cleft palate, asthma, recurrent sinopulmonary infections, attention deficit/hyperactivity disorder (ADHD), autism, and pervasive developmental disorder (PDD). Patient intermittently had abnormal TSH but never required treatment with levothyroxine and has not reported symptoms of hypo or hyperthyroidism. Patient medications through the period of evaluation include ibuprofen, lisdexamfetamine dimesylate, loratadine, albuterol, insulin pen needle, and ondansetron. Patient’s surgical history includes palatoplasty, adenoidectomy, and spinal fusion scoliosis repair.
Patient initially presented when 12 years old with concerns for micropenis, short stature, hypotonia, and weight gain. An hCG stimulation test to differentiate hypogonadotropic hypogonadism from expected low prepubertal gonadotropin levels was not initially done because the patient would not have tolerated the procedure. Patient was diagnosed with a severe hGH deficiency and early puberty and was subsequently treated with somatotropin for 1 year. The patient began treatment with somatotropin 2.4 mg once daily. The patient began a higher somatotropin dose of 2.6 mg once daily, began taking carotenall 2500 IU once daily, and began following a Mediterranean style diet following concerns for an elevated visceral adipose index. Patient discontinued somatotropin therapy following the achievement of a functional adult height and a normal IGF. Patient’s pubertal development was reported to be progressing, and pubertal delay was never initiated. At 20 years old, the patient’s testosterone, FSH/LH, cortisol, ACTH, prolactin, FT4, TSH, and IGF1 were retested to rescreen the patient for endocrine abnormalities and the patient had low testosterone level (214 ng/dL) alongside normal LH (4.4 mIU/mL) and FSH (4.1 mIU/mL) levels (Table 2).
Lab Values and Diagnostic Results for Caes Presentation #2.

Discussion
In our report, we present 2 cases of patients with 22q11.2 deletion syndrome with low levels of testosterone. The normal FSH and LH may indicate an inappropriate response to the low testosterone levels and is a failure to compensate. This is an example of secondary hypogonadism.
The exact mechanisms behind both primary and secondary etiologies for hypogonadism in patients with 22q11.2 deletion syndrome are not well understood. Perhaps, embryologic development of the hypothalamic–pituitary–gonadal (HPG) axis in patients with 22q11.2 variants may be aberrant, leading to decreased levels of gonadotropins. This may also lead to testicular dysfunction, leading to primary hypogonadism. 22q11.2 deletion syndrome is associated with other endocrine abnormalities such as hypoparathyroidism, hypocalcemia, and thyroid dysfunction, which may disrupt the HPG axis. Impaired SNAP29 function in these patients may also lead to decreased levels of testosterone due to impaired release at the level of the testes.
Cases of hypogonadism in patients with 22q11.2 are mostly unreported in the literature. The only other reported patient with hypogonadism and 22q11.2 deletion syndrome had diabetes, obesity, chronic kidney disease, and a pituitary adenoma, all factors that may explain hypogonadism. In both of our cases a thorough workup was conducted to assess for these etiologies of the hypogonadism. The workup was largely unremarkable, including an assessment for pituitary dysfunction and hemochromatosis. Additionally, head trauma may cause hypogonadism, but this was not present in our patient.
Interestingly these patients were both unexpectedly found to have low testosterone levels. This indicates that patients with 22q11.2 deletion syndrome and hypogonadism may often be underdiagnosed as there are no specific protocols in place to screen these patients. We propose that patients with 22q11.2 deletion syndrome receive testosterone, FSH, and LH levels to further identify cases of hypogonadism in these patients.
There are various limitations in our study. We include only 2 cases to assess the relationship between 22q11.2 deletion syndrome and hypogonadism. Further studies assessing testosterone, FSH, and LH levels need to be conducted in patients with 22q11.2 deletion syndrome. Additionally, our study does not include a long-term follow-up of these patients. It is unclear whether the hypogonadism is a long-term problem or a transient lab abnormality in patients with 22q11.2 deletion syndrome.
Conclusion
Further study is warranted to clarify the relationship between 22q11.2 deletion syndrome and hypogonadism. This can be done by obtaining testosterone, LH, and FSH levels in this patient population. Laboratory studies may also be done to clarify the relationship between SNAP29 and hypogonadism in these patients. Once the relationship between hypogonadism and 22q11.2 deletion syndrome is further clarified, hopefully the clinical guidelines will be updated to reflect routine screening in these patients for hypogonadism.
Footnotes
Acknowledgements
We would like to thank the patients for consenting for their information to be included in this case series.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent for Publication
Verbal informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Prior Presentation of Abstract Statement
Abstract has not been previously presented.