
Abstract
Rosai–Dorfman disease (RDD) is a rare non-Langerhansian histiocytosis, classically manifesting as massive cervical lymphadenopathy. Isolated extra-nodal forms, particularly in soft tissues, are exceptional and may simulate a malignant tumor. We report the case of a 56-year-old patient presenting with a painless mass of the right thigh that had been evolving for several weeks. Clinical examination revealed a deep mass measuring approximately 10 cm. Magnetic resonance imaging showed a tissue formation encompassing the femoral vessels, responsible for thrombosis with vascular stenosis. A surgical biopsy was performed. Histological analysis, coupled with immunohistochemistry, confirmed the diagnosis of RDD. Anticoagulant therapy with apixaban was instituted for the venous thrombosis. The evolution was marked by spontaneous regression of the mass without recourse to additional surgery. This case illustrates an atypical presentation of RDD through isolated involvement of the soft tissues of the thigh, complicated by deep venous thrombosis. Diagnosis is based on histopathological examination. Treatment is individualized and may be limited to monitoring in nonprogressive forms. RDD must be included in the differential diagnosis of deep soft tissue masses. Early recognition can avoid invasive treatment.
Keywords
Introduction
Rosai–Dorfman disease (RDD) is a rare, benign phagocytic histiocytosis that usually presents as massive, painless lymphadenopathy. 1 Although the classic presentation involves bilateral cervical lymphadenopathy, often with fever, this entity can rarely manifest in an extra-cervical form, or even exclusively in extra-nodal sites, making its diagnosis more challenging. 2 Extra-nodal localization account for less than 30% of cases, with a predilection for the skin, upper respiratory tract, central nervous system, and gastrointestinal tract. Isolated presentation touching the soft tissues of the limbs is exceptional. 3 We herein report a case of RDD presenting as a painless mass of the left thigh, highlighting the diagnostic challenges and therapeutic options in this atypical context.
Case Presentation
A 56-year-old man with a medical history of hypertension, type 2 diabetes managed with oral hypoglycemic agents, and a 20-pack-year smoking habit was referred for evaluation of a progressively enlarging, painless mass in his right proximal thigh, evolving over 6 months. He reported no constitutional symptoms such as fever, weight loss, or functional impairment. Clinical examination revealed a hemodynamically stable patient in good general condition, with a firm, deeply fixed 10 cm mass at the root of the right thigh. No palpable lympohadenopathy was found. Magnetic resonance imaging (MRI) identified a tumor-like lesion originating from the right femoral, Sartorius, and adductor longus muscles, encasing the femoral vascular bundle (Figure 1). Subsequent CT angiography further delineated the mass (74 × 60 × 119 mm) located inter-aponeurotically between the quadriceps and adductor muscles, compressing the right superficial femoral vein (causing complete thrombosis) and arteries (resulting in moderate stenosis). Additionally, diffuse atherosclerotic mediacalcosis of the abdominal aorta and lower limb arteries was noted, alongside multifocal severe stenoses (70–90%) in bilateral superficial femoral arteries. A smaller contra lateral mass (38 × 33 × 61 mm) was observed along the left femoral vasculature. Thoracoabdominal CT ruled out metastatic disease. Laboratory findings included mild leukocytosis (12,500 cells/mm³), thrombocytosis (439,000 cells/mm³), and elevated C-reactive protein (75 mg/L).
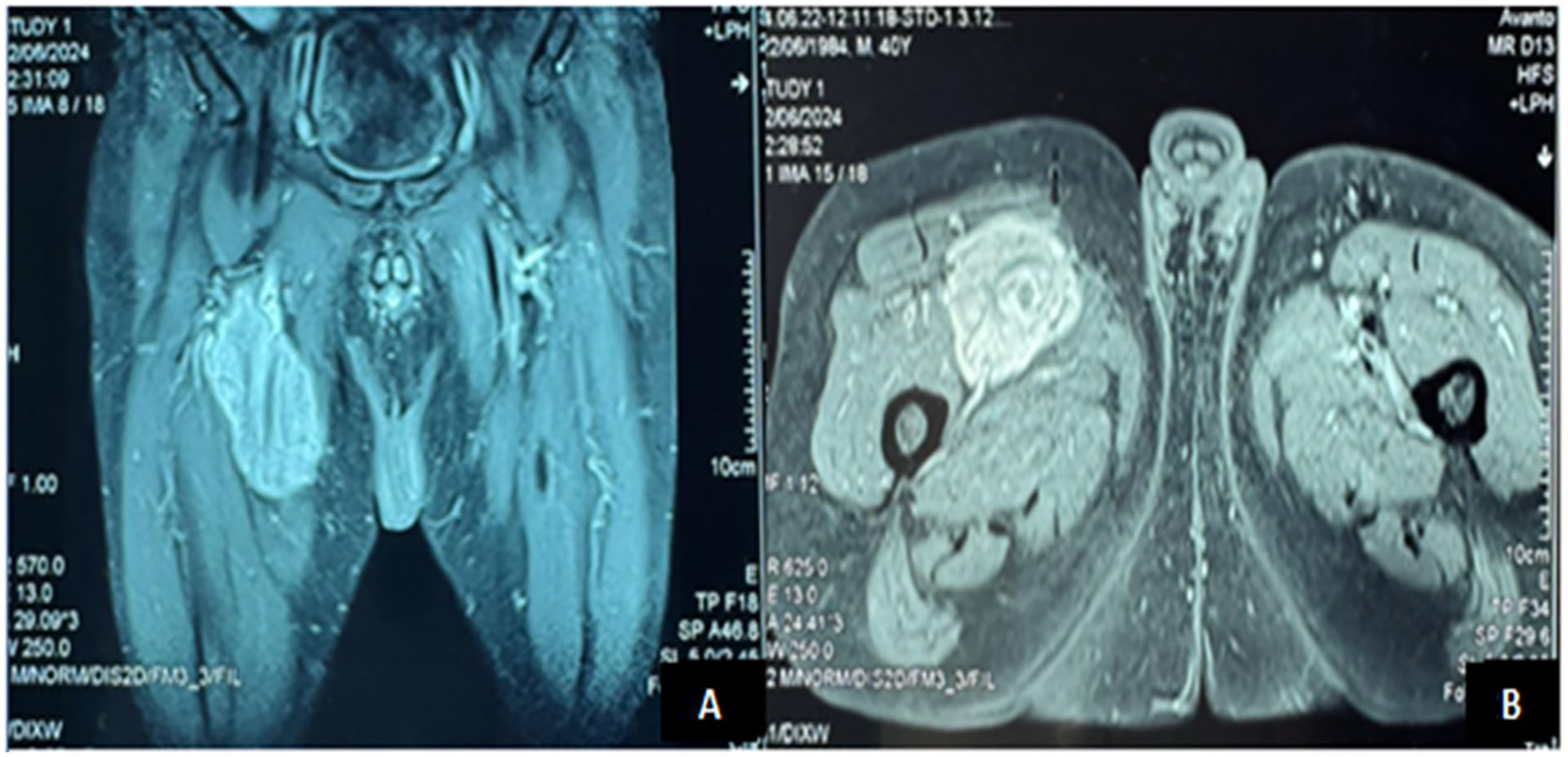
MRI images showing the tumor-like lesion originating from the right femoral, Sartorius, and adductor longus muscles. (A) Coronal section. (B) Axial Section.
Initial ultrasound-guided biopsy revealed nonspecific fibro inflammatory tissue with scattered lymphocytes and plasma cells. Due to the mass’s fixation and diagnostic uncertainty, a surgical biopsy under spinal anesthesia was performed. Intraoperative exploration identified whitish superficial tissue, with frozen section analysis suggesting inflammatory changes. Deeper sampling yielded grayish tissue, which on final histopathology demonstrated fibroadipose stroma infiltrated by sheets of histiocytes exhibiting rounded nuclei, eosinophilic or foamy cytoplasm, and emperipolesis (intact lymphocytes within histiocytic cytoplasm). Accompanying features included plasma cells, neutrophils, and lymphoid nodules (Figure 2). Immunohistochemistry confirmed histiocytic positivity for S100 (intense, diffuse), CD68, and cyclin D1, while markers for malignancy (MDM2, CD34) and other histiocytic disorders (ALK, CD1a) were negative. The histopathological features, including emperipolesis and S100/CD68-positive histiocytes, combined with the immunohistochemical profile, definitively established the diagnosis of RDD (Figure 3). KRAS and MAP2K1 mutations were not tested in our patient’s case due to a lack of resources, considering that we are based in a low-income country, where these tests are prohibitively expensive and not available to all institutions.

(2) Rosai-Dorfman disease. Prominent histiocytic infiltrate in a background of inflammatory cells (Ax200). (2) Histiocytes have an enlarged, round to oval hyperchromatic nuclei and an abundant eosinophilic cytoplasm (Bx400).
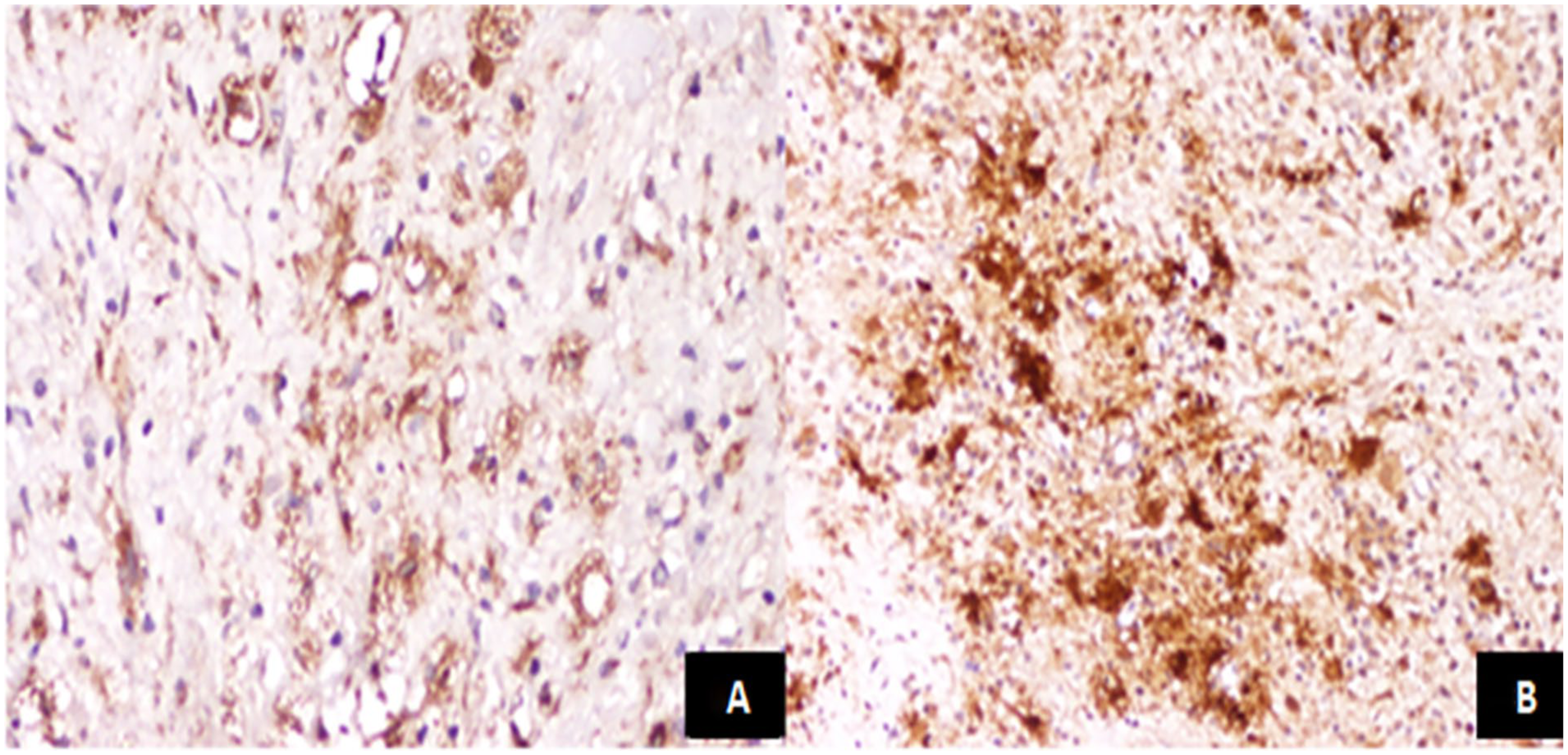
Histiocytic infiltrate highlighted by positive staining for CD68 (Ax200) (3A) and S100 (Bx100) (3B).
The patient was initiated on apixaban for femoral vein thrombosis. Remarkably, the mass regressed spontaneously by 50% (from 10 cm to 5 cm) within three months. At 6-month follow-up, he remained asymptomatic with no disease progression. Given the vascular proximity and favorable clinical course, surveillance was elected over surgical intervention.
Discussion
RDD, also known as sinus histiocytosis with massive lymphadenopathy, is a rare condition of undetermined etiology, belonging to the group of non-Langerhansian histiocytoses. It was first described in 1969 by Rosai and Dorfman. 4 The exact incidence of this condition remains poorly known due to its rarity, although a few hundred cases have been reported in the literature. It mainly affects children and young adults, with peak incidence between the second and third decades of life. A sex ratio slightly in favor of men has been observed, estimated at around 1.4:1. 5
The classic presentation is massive, often bilateral, cervical lymphadenopathy, sometimes associated with systemic signs such as fever, asthenia, weight loss or hyperleukocytosis. 6 In around 40% of cases, extra-ganglionic involvement is reported, affecting various organs: skin, respiratory tract, central nervous system, orbits, bone, kidney or soft tissue. Isolated soft-tissue involvement, particularly in the limbs, is exceptional and can lead to misdiagnosis. 7 Our case illustrates this atypical presentation. This type of isolated soft-tissue localization, without lymph node involvement, is very rare and often confused with other benign or malignant soft-tissue tumors. Complementary examinations, notably MRI, may be useful to characterize the lesion, assess its anatomical position and guide surgical management.8,9 However, imaging remains nonspecific. Biological tests are often normal or of limited contribution and do not allow us to orientate the diagnosis with certainty. Pre-operative or pre-biopsy diagnosis is therefore very difficult. 10 Diagnostic confirmation relies exclusively on histopathological analysis of the surgical excision. It reveals infiltration by large histiocytes with abundant cytoplasm, sometimes containing intact cells (lymphocytes, plasma cells)—a phenomenon known as emperipolesis, which is characteristic but not pathognomonic.11–13 Immunohistochemical study confirms the diagnosis: histiocytes express the markers S100 and CD68, while being negative for CD1a, thus ruling out Langerhans histiocytosis. 13 In the absence of a formal therapeutic consensus, strategy depends on clinical presentation. In symptomatic localized forms, complete surgical resection is often the preferred treatment, and in most cases is both sufficient and curative.2,13 Corticosteroid therapy, immunotherapy or chemotherapy may be considered in diffuse, recurrent or life-threatening forms, although these indications remain rare. 14 Disease outcome is generally favorable. In the majority of cases, spontaneous or postsurgical regression is observed. Local recurrence is possible, but infrequent. The overall prognosis is excellent, with long-term survival almost equivalent to that of the general population, in the absence of severe systemic involvement.3,5 The pathophysiology and pathogenesis of RDD remain unknown. Infection with human herpesvirus 6, Epstein–Barr virus and parvovirus B19 have been suspected as triggering events. 15 Several genetic mutations have also been identified recently, including mutations in the ARAF, MAP2K1, NRAS and KRAS genes.16,17 KRAS and MAP2K1 are associated with 33% of RDD cases, suggesting that the RASMAP2K1 pathway may be an integral component in the development of the disease and therefore a potential therapeutic target. Thus, although rare, RDD should be considered in the differential diagnosis of soft tissue masses, even in the absence of lymph node involvement, to avoid inappropriate management and preserve a good prognosis.
Conclusion
RDD is a rare entity, classically characterized by massive cervical lymph node involvement, but can also manifest itself in isolated, sometimes puzzling, extra-ganglionic forms. Our case highlights the possibility of an unusual localization in the soft tissues of the thigh, in the absence of any associated systemic or lymph node signs. This atypical presentation calls for a rigorous diagnostic approach, in which histopathological and immunohistochemical analysis remains the key tool for making the diagnosis. Complete surgical resection is an effective treatment option in localized forms. This case reminds us of the importance of keeping this pathology in mind in the differential diagnosis of soft tissue masses, in order to avoid over-treatment and ensure appropriate management.
Footnotes
Acknowledgements
We acknowledge the support of our colleagues and our institution throughout the development of this work.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Informed Consent
Verbal informed consent was obtained from the patient for their anonymized information to be published in this article.