
Abstract
Neonatal diabetes mellitus (NDM) is a rare metabolic disorder that develops within the first 6 months of life and can have a wide clinical presentation which includes diabetic ketoacidosis (DKA). We describe a 56-day-old female with permanent NDM whose clinical presentation included polyuria, fever, vomiting, and dehydration. Laboratory workup indicated DKA. The patient started her treatment with subcutaneous insulin, but her blood sugar level was poorly controlled. She was later found to have a KCNJ11 mutation and was subsequently switched to sulfonylurea, which offered better control of blood sugars. Our case highlights the importance of recognizing signs and symptoms such as polyuria and vomiting along with the profound impact of genetic changes such as KCNJ11 in the pathophysiology of the condition. Genetic counseling is necessary for affected families, and increases awareness of potential complications, particularly those related to DKA and associated neurological risks.
Introduction
Permanent neonatal diabetes mellitus (PNDM) is a rare form of diabetes with a lifetime duration, which develops within the first 6 months of life. Its incidence ranges from 1 in 89 000 to 400 000 births. 1 It mostly results from mutations in KCNJ11 and ABCC8 genes which form the subunits of the ATP-sensitive potassium (K-ATP) channels located in pancreatic β cells. These mutations prevent channel closure in response to glucose, impairing insulin secretion, and leading to hyperglycemia. 2 These mutations have a strong impact on monogenic diabetes because its origin determines the prognosis, treatment, and inheritance. In these kinds of cases, insulin therapy can generally be replaced by oral sulfonylurea medication, leading to better glucose levels, and improved neurological outcomes. 3
We reported a 56-day-old female patient who presented with diabetic ketoacidosis (DKA) symptoms and was diagnosed with PNDM due to a heterozygous pathogenic KCNJ11 gene mutation and was treated with sulfonylurea with more stable glycemic readings than her reading when she started on insulin.
Case Presentation
A 56-day-old female infant born to nonconsanguineous parents via normal vaginal delivery with a birth weight of 2.7 kg, presented with a fever of 39.5 °C alleviated by antipyretic medication, a single episode of vomiting large amounts of milky content, a 1-week history of increased feeding, and a 3-week history of polyuria. There were no signs of poor feeding or inadequate weight gain.
By examination, the patient appeared ill, dehydrated, and hypoactive, exhibiting respiratory distress with subcostal retractions, and the neurological exam was unremarkable. She had a weight of 3.7 kg, a head circumference of 37.5 cm, and a length of 58 cm. Her vital signs were unstable, with a blood pressure of 100/55 mmHg, a respiratory rate of 55 bpm, a pulse rate of 179 bpm, and a temperature of 35 °C. Laboratory investigations revealed a random blood sugar (RBS) level of 636 mg/dL, an HbA1c of 11.6%, low insulin levels, and a low C-peptide level of 0.9 ng/mL; other labs in the hospital are shown in Table 1. An abdominal ultrasound done to rule out structural abnormalities showed a normal-sized pancreas with no other abnormalities.
Comprehensive Laboratory Results for the DKA Patient Indicate Hyperglycemic Metabolic Acidosis.
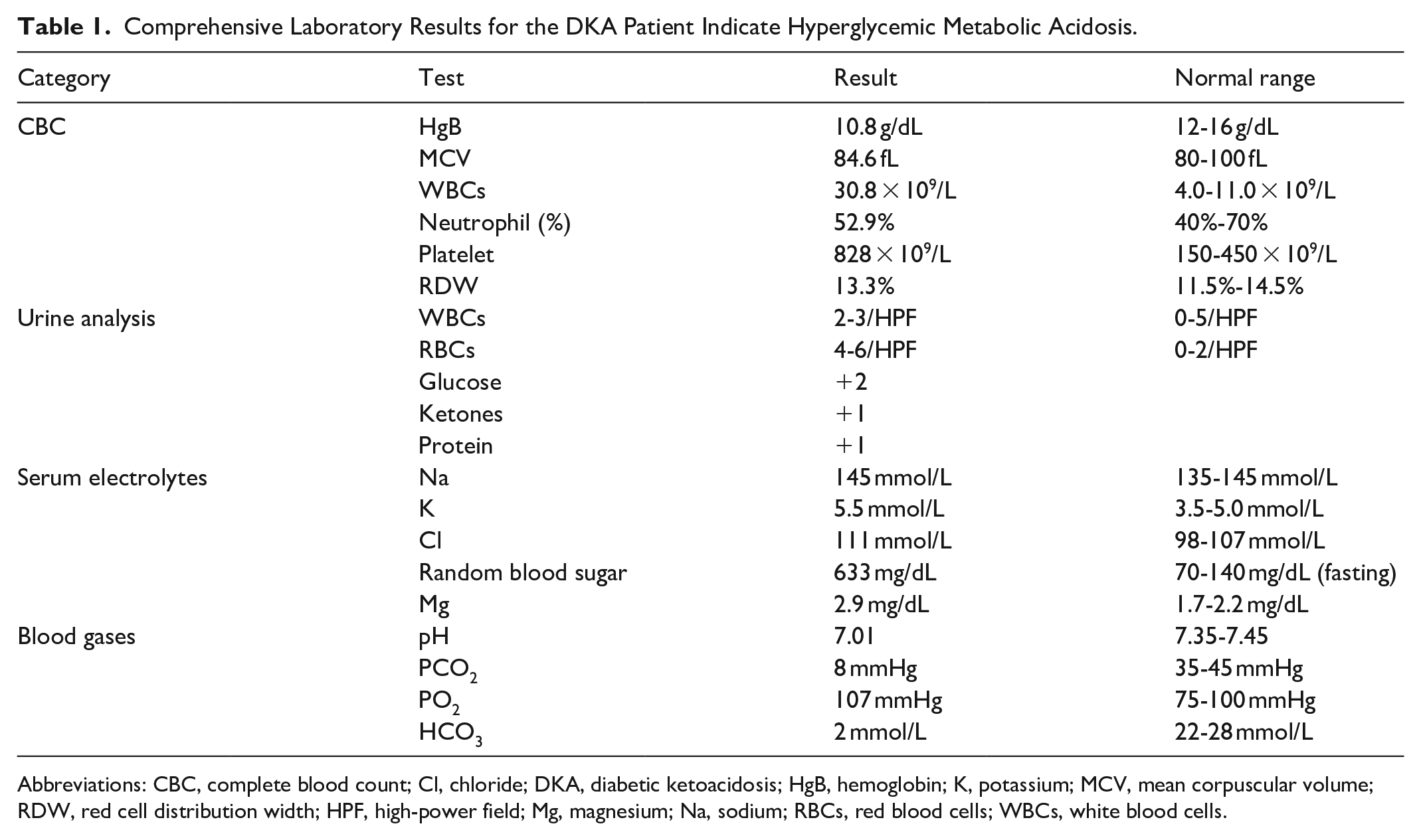
Abbreviations: CBC, complete blood count; Cl, chloride; DKA, diabetic ketoacidosis; HgB, hemoglobin; K, potassium; MCV, mean corpuscular volume; RDW, red cell distribution width; HPF, high-power field; Mg, magnesium; Na, sodium; RBCs, red blood cells; WBCs, white blood cells.
The patient was initially treated with intravenous ampicillin and cefotaxime as a case of potential sepsis due to meningitis. A cerebrospinal fluid culture was negative, with protein levels within normal limits, a glucose concentration of 59 mg/dL, and a white blood cell count of 157 cells/µL. Her blood gases showed metabolic acidosis with a pH level of 7.01, HCO3− of 2 mEq/L, PaO2 of 107 mmHg, and PaCO2 of 8 mmHg. Along with her random blood sugar (RBS) levels, glucosuria and ketonuria, she was diagnosed with DKA and was started on an insulin drip following the DKA protocol. She also received intravenous normal saline. Her condition improved, and she was transitioned to subcutaneous insulin therapy with insulin detemir (Levemir), and her last RBS was 250 mg/dL. Upon discharge, she was prescribed 0.5 to 1 IU of Actrapid and 1 to 2 IU of Levemir according to glucose level, with regular blood glucose monitoring.
Genetic testing identified a heterozygous missense variant in the KCNJ11 gene on exon 1 p.(ser3Cys). A genetic test was also done for the parents, and KCNJ11 was not detected in the parent’s sample, so the variant raised is de novo.
Due to her instability of her glucose readings, the patient was readmitted with similar symptoms. Subcutaneous insulin therapy was discontinued, and oral sulfonylurea was initiated at a dose of 0.2 mg/kg/day, increasing by 0.2 mg/kg/day until a maximum dose of 1 mg/kg/day was reached, based on fasting blood glucose levels. The patient experienced hypoglycemia with a blood glucose reading of 55 mg/dL at the maximum dose, prompting a reduction in the dose to 0.6 mg/kg/day administered twice daily. Blood glucose levels stabilized, and she was discharged on oral glibenclamide 1.5 mg twice daily.
The patient is currently under regular follow-up with a pediatric endocrinologist, demonstrating stable blood glucose levels and normal growth and development (Figure 1).

Comparison of serum random glucose levels before (on insulin) and after (on sulfonylurea) therapy.
Discussion
Neonatal diabetes mellitus (NDM) is a rare, monogenic disorder that presents within the first 6 months of life. NDM is associated with more than 20 genes, with the most common being KCNJ11, INS, 6q24, and ABCC8. 4 NDM is categorized into 2 types, which are transient NDM (TNDM) and PNDM. TNDM is an exceedingly rare genetic disorder (1 in 90 000-160 000 live births) caused by methylation changes in PLAGL1 and HYMAI that result in insulin-dependent hyperglycemia. Over 95% of cases present with intrauterine growth restriction, and the majority of symptoms resolve within 12 weeks.1,5
PNDM is another form of NDM that presents in infancy (1 in 89 000-400 000 live births) and is defined by persistent hyperglycemia and insulin dependence in 40% to 50% of cases. It is frequently associated with KCNJ11 and ABCC8 mutations. 1 In this case, ABCC8, KCNJ11, INS, and EIF2AK3 mutations were examined. Genetic testing showed a result of heterozygous mutation in the KCNJ11, a missense variant (p.Ser3Cys) described at c.8C>G.
Mutations in the KCNJ11 gene are one of the most prevalent causes of PNDM, accounting for 30% of the cases. 6 KCNJ11 mutations are typically de novo in 80% of cases, while the remaining cases are inherited in an autosomal dominant pattern. NDM with these mutations typically presents from birth to 26 weeks of age, with an average onset at 5 weeks. In our case, the patient presented at the age of 8 weeks. 7
NDM can present with varying signs and symptoms, which may not always appear immediately after birth. Symptoms may be insidious, including polyuria, polydipsia, or failure to thrive. In some cases, presentation can be acute, with DKA or altered mental status, or it may be asymptomatic and detected incidentally. The likelihood of DKA increases with age due to challenges in recognizing early signs in infants. 8 In our case, the patient presented with polyuria and vomiting; labs and blood gases showed metabolic acidosis with high blood glucose, and urine analysis showed glycosuria and ketonuria which confirmed the diagnosis of DKA.
NDM is also reported to present with a wide range of clinical features, such as hyperglycemic hyperosmolar state, and intracranial hemorrhage. Additionally, it can be part of complex syndromes such as developmental delay, epilepsy, and neonatal diabetes syndrome. These diverse and potentially fatal presentations emphasize the critical need for early diagnosis and tailored treatment. Selected case reports illustrating these complications are summarized in Table 2.9-11
Reported Case Reports Highlighting Severe and Atypical Presentations of Neonatal Diabetes Mellitus.

Abbreviations: DEND, developmental delay, epilepsy, and neonatal diabetes; DKA, diabetic ketoacidosis; HHS, hyperosmolar state; ICH, intracranial hemorrhage; NDM, neonatal diabetes mellitus.
Our case stands out due to the presence of a rarely reported mutation (p.Ser3Cys), severe DKA at presentation, absence of neurological complications, and excellent response to early sulfonylurea therapy.
The treatment of NDM depends on its genetic cause. For patients with mutations in the KCNJ11 or ABCC8 genes, which affect the Kir6.2 and SUR1 subunits of the K-ATP channel, sulfonylureas are a highly effective option. These medications work by closing the K-ATP channels, stimulating insulin secretion, and improving blood sugar control. 8 Sulfonylureas are especially beneficial because they are taken orally, avoiding the need for insulin injections, and often achieving stable glucose levels, as treatment with insulin is difficult to manage due to the low weight of these infants. The therapeutic margins between hypoglycemia and hyperglycemia are very narrow and are harmful to the neurological development of the infant. Our patient was treated with subcutaneous insulin initially before the genetic test result was confirmed for the KCNJ11 mutation, and his readings were fluctuating and not controlled, so then, after confirming the mutation, he was transitioned to sulfonylurea (Glibenclamide) with more controlled readings.
Early genetic testing is important to identify the underlying cause and choose the best treatment, ensuring better outcomes for patients. It also helps predict associated conditions, making it recommended for all suspected cases, especially in families with diabetes in children under 12 months. 12 Also, it is good for avoiding the reported complications associated with PNDM, such as Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX syndrome), Wolcott–Rallison syndrome, and Wolfram syndrome. 1 In our case, the patient had a de novo mutation, and the family was advised that future offspring have a 50% risk of inheriting the variant and developing NDM.
Conclusion
This case reports a 56-day-old female infant who presented with DKA symptoms. She was initially treated with insulin without controlled reading. After conducting the genetic test, which confirmed the KCN11 mutation, she transitioned to sulfonylurea with more controlled glycemic readings. This case emphasizes the importance of genetic counseling for families to comprehend the condition’s heritability. Future advancements in genetic testing and personalized medicine offer potential for enhanced management of NDM.
Footnotes
Acknowledgements
The authors express profound gratitude to the Polytechnic Medical Students’ Research Association (PMRA) for their invaluable contributions and unwavering support that significantly enriched every stage of the research journey. Also, the authors express their gratitude to the patient’s family for their great contribution.
Ethical Considerations
This case report is exempt from ethical approval in our institute, Palestine Polytechnic University.
Consent for Publication
Written informed consent was obtained from the patient’s family for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data used to support the findings of this study are included in the article.