
Abstract
Mature plasmacytoid dendritic cell proliferation (MPDCP) is a rare, clonal but nonmalignant entity often associated with myeloid neoplasms such as chronic myelomonocytic leukemia (CMML), myelodysplastic syndromes, and acute myeloid leukemia. While typically confined to the bone marrow, nodal MPDCP is exceedingly rare and may mimic blastic plasmacytoid dendritic cell neoplasm (BPDCN), posing diagnostic challenges. We report a 78-year-old male with CMML-1 and progressive cervical lymphadenopathy. Workup revealed monocytosis, ASXL1 and CBL mutations, and CMML. Lymph node biopsy showed paracortical expansion by small mononuclear cells with plasmacytoid features. Immunophenotyping identified a CD4+, CD123+, CD303+, HLA-DR+, lysozyme+, CD56− population, consistent with MPDCP. A subset expressed TdT and granzyme B, with a Ki-67 index of 20% to 30%. Next-generation sequencing confirmed the same ASXL1 and CBL mutations in the lymph node, supporting clonal relation to CMML. Key differential diagnoses included BPDCN, T-cell lymphomas, Langerhans cell histiocytosis, and Kikuchi–Fujimoto disease. Absence of CD56, mature cytomorphology, and molecular concordance favored MPDCP. This case highlights the importance of distinguishing nodal MPDCP from malignant mimics. MPDCP may reflect immune evasion, altered cytokine signaling, or clonal progression in myeloid neoplasms. The patient was initially treated with hydroxyurea, later transitioned to decitabine/cedazuridine (Inqovi) for disease progression. Follow-up marrow biopsy showed stable CMML-2 with persistent mutations, and the patient remains under close monitoring. Recognizing MPDCP in unusual locations is critical for accurate diagnosis and prognostication. Further studies are warranted to clarify its molecular pathogenesis and potential as a biomarker of disease evolution in CMML and related disorders.
Keywords
Introduction
Plasmacytoid dendritic cells (pDCs) are specialized antigen-presenting cells that play a vital role in innate immunity, particularly against viral infections by producing high levels of type I interferons.1,2 They originate from bone marrow hematopoietic stem cells and differentiate from both myeloid and lymphoid progenitors, resulting in a small fraction of peripheral blood mononuclear cells. Under normal physiological conditions, pDCs are present in low numbers in the blood, bone marrow, and lymphoid tissues. 3
pDC expansions are classified into 3 categories:
MPDCP is characterized by a proliferation of morphologically mature pDCs that express CD4, CD123, and CD303, typically with a low proliferative index (Ki-67 <10%). Unlike BPDCN, MPDCP usually lacks CD56 expression, which aids in its distinction.5,6,9,12,13
MPDCP is most identified in bone marrow samples of patients with myeloid neoplasms, reported in ~20% to 30% of such cases.11,14 However, nodal MPDCP remains exceedingly rare, with only a few cases described in the literature. This condition involves the proliferation of mature pDCs within lymph nodes, which can mimic lymphomatous or leukemic processes, posing significant diagnostic challenges.
For instance, a recent case involved a patient with CALR-mutated myelofibrosis who developed generalized lymphadenopathy; histologic examination revealed extensive pDC proliferation, initially concerning for lymphoma. However, immunophenotyping confirmed nodal MPDCP. 15 Another report described a patient with chronic myelomonocytic leukemia (CMML) presenting with marked nodal pDC proliferation, which was associated with adverse prognosis and progression to acute myeloid leukemia (AML). 16
The underlying molecular mechanisms of MPDCP remain under investigation. Recent studies have elucidated that MPDCPs associated with myeloid neoplasms are clonally related to the underlying myeloid neoplasm. This clonal relationship is evidenced by shared genetic mutations such as ASXL1, TET2, or SRSF2, between the proliferating pDCs and the malignant myeloid cells.
In CMML, pDCs have been found to harbor mutations in genes mirroring those present in the monocytes of the same patients. Similarly, in AML, pDCs, and leukemic blasts have been shown to share chromosomal abnormalities, including deletions and translocations, further supporting their clonal relationship. 17
Here, we present a rare case of nodal MPDCP in a patient with CMML, emphasizing the diagnostic complexity, molecular characteristics, and clinical implications of this entity. We also explore the importance of distinguishing nodal MPDCP from BPDCN and other mimickers, highlighting the need for integrated morphologic, immunophenotypic, and molecular analysis in hematopathologic evaluation.
Case Presentation
A 78-year-old male with a known history of CMML-1 presented with progressive cervical lymphadenopathy. He was initially found to have monocytosis in September 2021 and was formally diagnosed with CMML several months later. At the time of diagnosis, laboratory studies revealed a white blood cell (WBC) count of 12.75 × 109/L with 20% monocytosis, hemoglobin of 13.4 g/dL, and a platelet count was 257 000/µL. Next-generation sequencing (NGS) identified mutations in ASXL1 (variant allele frequency [VAF] 47%) and CBL (VAF 60%). Cytogenetics analysis of the bone marrow aspirate showed a normal karyotype (46, XY).
On physical examination, a firm, nontender cervical lymph node was noted, without evidence of hepatosplenomegaly. An excisional biopsy of the lymph node was subsequently performed.
Histopathologic sections revealed a partially preserved nodal architecture with expanded paracortical areas with an infiltrate of small mononuclear cells with pale cytoplasm and slightly dispersed chromatin (Figure 1).

Histologic examination of the lymph node excisional biopsy (hematoxylin and eosin stain (A) ×20 magnification and (B) ×400 magnification) demonstrates a partially preserved architecture, with paracortical expansion by a proliferation of small mononuclear cells with moderate amounts of pale cytoplasm and slightly dispersed chromatin.
Immunohistochemical and flow cytometric analyses (Figures 2 and 3) demonstrated that these atypical mononuclear cells had a CD45-dim immunophenotype and expressed CD4, CD123, CD2 (partial), CD5, CD10 (partial), CD33, CD36, CD38, HLA-DR, CD68 (KP1), CD117, TCL1a, and granzyme B. A subset of these cells also expressed TdT. The atypical population was negative for CD3, CD8, CD7, CD13, CD34, CD19, CD20, CD64, myeloperoxidase, CD25, CD79a, CD11c, MUM1, and kappa/lambda light chains. CD14 highlighted only scattered positive cells. Ki-67 staining showed a proliferation index of ~20% to 30% within the infiltrate. Despite the expression of TdT and granzyme B, the cytologic features were relatively bland, with absence of CD56 expression and a moderate proliferation index. These findings supported a diagnosis of MPDCP in the setting of CMML.
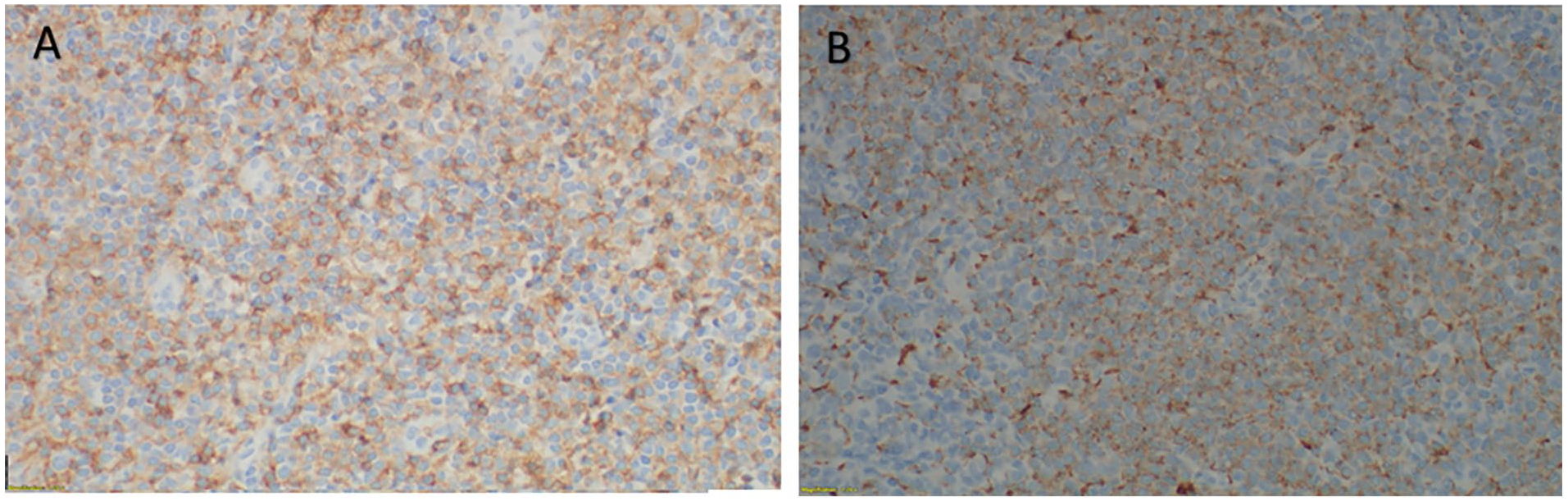
Immunohistochemical stains show that those atypical mononuclear cells (plasmacytoid dendritic cells) are positive for (A) CD4 and (B) CD68 antibodies.
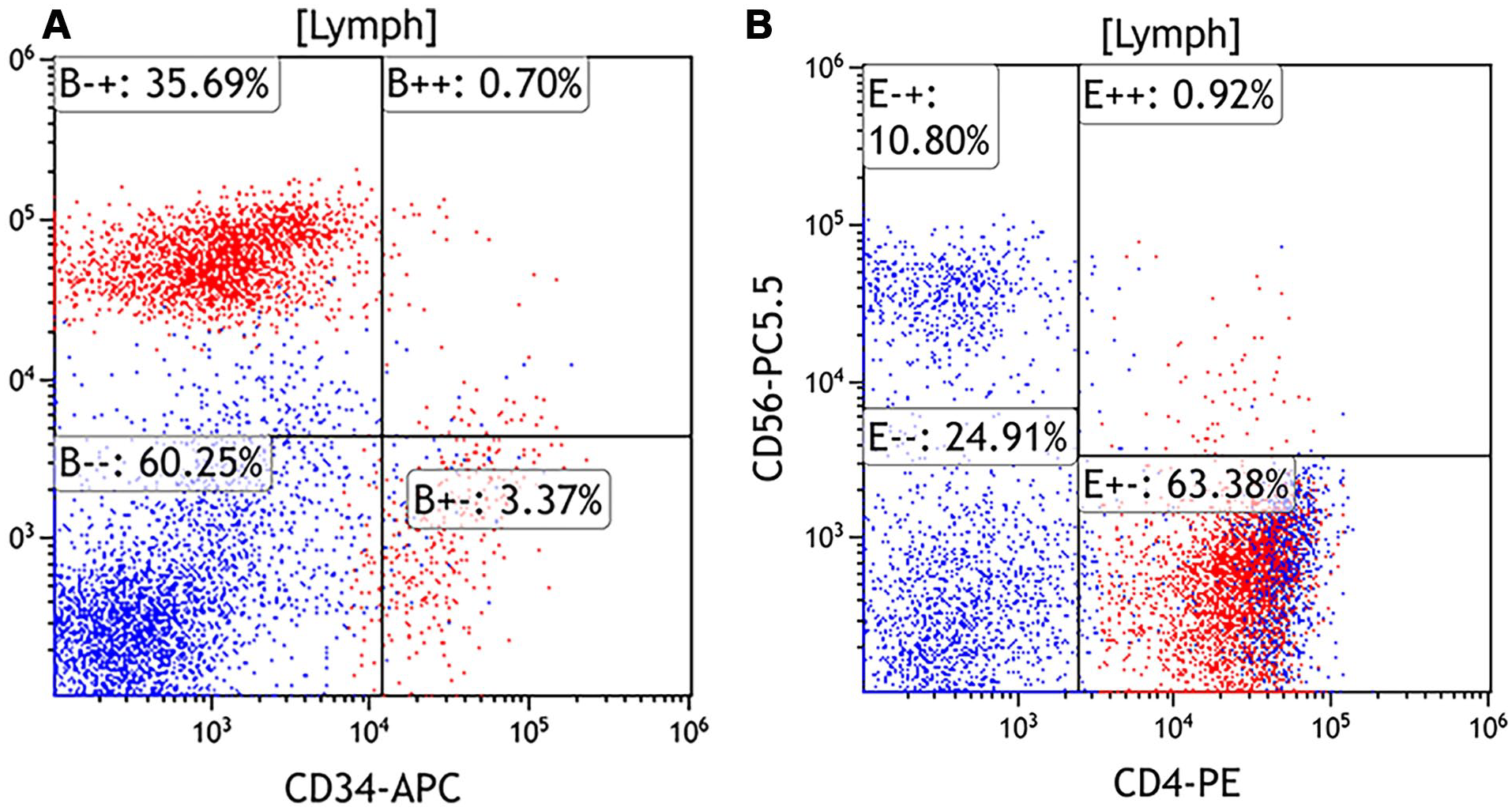
Flow cytometric immunophenotypic analysis demonstrates an atypical CD45—dim population (red population) in the lymphocytes gate (A) this population is CD34−/CD123+ and (B) CD4+/CD56−.
A concurrent bone marrow biopsy confirmed CMML-2 with 5% blasts and persistent ASXL1 and CBL mutations. NGS performed on the lymph node also revealed identical mutations, confirming clonal relationship between the MPDCP and the underlying myeloid neoplasm.
Several months later, the patient experienced leukocytosis, prompting initiation of hydroxyurea, which reduced the WBC count to 10.0 × 109/L. Repeat NGS confirmed persistence of ASXL1 and CBL mutations without emergence of FLT3 mutations. Due to disease progression, treatment was escalated to Inqovi (decitabine/cedazuridine) (Taiho Oncology, Inc.), based on disease burden and patient tolerance.
A follow-up bone marrow biopsy showed persistent CMML-2 with 5% blasts and grade 1 reticulin fibrosis (MF-1). Recent laboratory evaluation demonstrated relatively stable disease, with hemoglobin at ~8.1 g/dL, WBC counts ranging from 11.4 to 13.4 × 109/L, and platelets ranging from 97 000 to 133 000/µL. The patient continues treatment with Inqovi and remains under active hematologic monitoring.
Discussion
This case report presents a rare instance of nodal MPDCP in a patient with CMML-1. MPDCP, though uncommon, is primarily associated with myeloid neoplasms, including CMML, AML, and myelodysplastic syndromes (MDS), and is reported in ~20% to 30% of bone marrow samples in such contexts.5 -10
This case highlights the diagnostic challenge of distinguishing MPDCP from other hematologic malignancies, particularly BPDCN, a rare and aggressive neoplasm. Accurate differentiation is essential, as it significantly influences prognosis and treatment planning.11,18
Key distinguishing features lie in cytomorphology, immunophenotype, and clinical behavior.
MPDCP is composed of mature-appearing pDCs with round to oval nuclei, finely dispersed chromatin, minimal cytoplasm, and a low proliferation index, in contrast, BPDCN cells exhibits a blastic morphology with higher nuclear-to-cytoplasmic ratios, irregular nuclear contours, and prominent nucleoli. 6
Immunophenotypically, both MPDCP and BPDCN express CD4, CD123, and HLA-DR. However, CD56 expression—a hallmark of BPDCN—is characteristically absent in MPDCP. 13 Additional markers such as lysozyme and mature myeloid antigens (CD11c, CD13, CD33) may aid in further classification.
Given the overlap in morphology and immunophenotype, comprehensive pathologic evaluation is critical (Table 1) for an accurate diagnosis, as the management and prognosis of MPDCP differ significantly from that of BPDCN, with BPDCN often associated with systemic symptoms such as fever, weight loss, and cytopenias6,19 and requiring more aggressive systemic therapies. 20 Notably, while our case showed atypical features, such as the relatively elevated Ki-67 and partial TdT expression, are atypical for MPDCP. These findings expand the spectrum of MPDCP but do not the overall diagnosis of MPDCP due to the absence of CD56 and other blastic features.
Distinguishing Features Between MPDCP and BPDCN.

Abbreviations: AML, acute myeloid leukemia; BPDCN, blastic plasmacytoid dendritic cell neoplasm; CMML, chronic myelomonocytic leukemia; MDS, myelodysplastic syndromes; MPDCP, mature plasmacytoid dendritic cell proliferation; pDCs, plasmacytoid dendritic cells.
Molecular Pathogenesis and Clonality of MPDCP in Myeloid Neoplasms
Molecular studies suggest that MPDCP in myeloid neoplasms represents a clonal expansion that shares genetic mutations with the underlying malignant hematopoietic clone.
In AML with pDC proliferation (pDC-AML), RUNX1 mutations are observed in over 70% of cases, significantly more than in AML without pDC involvement, reinforcing the clonal relationship between pDCs and leukemic blasts. Similarly, in CMML with pDC expansion (pDC-CMML), mutations in genes such as TET2, SRSF2, and ASXL1, common drivers in myeloid neoplasia, are consistently identified in both pDCs and the myeloid component, further supporting their shared origin.7,17 Understanding this shared mutational landscape not only aids in accurate diagnosis but also presents opportunities for targeted therapeutic intervention.
Our patient’s lymph node and bone marrow samples both harbored ASXL1 and CBL mutations, reinforcing the clonal relationship between the MPDCP and the underlying CMML. ASXL1 mutations, seen in ~45% of CMML cases, are associated with poor overall survival and disease progression. The presence of these mutations in proliferating pDCs suggests that MPDCP may serve as a biomarker for clonal evolution and an increased risk of transformation to AML.21,22
Prognostic Implications
While MPDCP is generally considered nonmalignant, its presence in myeloid neoplasms, particularly in CMML—may be associated with adverse prognosis. Proliferating clonal pDCs, often linked to RAS pathway activation, may contribute to leukemic evolution and reduced leukemia-free survival. 21 Proposed mechanisms include immunosuppressive microenvironment development, increased regulatory T-cell activity, and the expression of immunoregulatory enzymes such as indoleamine 2,3-dioxygenase by pDC aggregates, all of which may facilitate disease progression. 23
MPDCP associated with AML appears to carry a more aggressive clinical course compared to MPDCP in CMML or MDS. 24 These observations highlight the importance of evaluating MPDCP within the clinical and genetic context of the underlying myeloid neoplasm.
Aberrant activation of the JAK-STAT signaling pathway and dysregulated cytokine signaling have also been implicated in pDC proliferation in CMML. JAK-STAT pathway dysregulation contributes to unchecked proliferation and impaired differentiation. Targeted therapies modulating this pathway are under active investigation.5,7,19,25
Differential Diagnosis
The differential diagnosis for MPDCP includes both neoplastic and reactive entities. Neoplastic mimics include Langerhans cell histiocytosis: expresses CD1a and langerin (CD207), which are absent in MPDCP. 26 T-cell lymphomas with pDC differentiation: often show T-cell receptor gene rearrangements, unlike MPDCP. 12
Reactive pDC expansions may be seen in: SLE, Kikuchi–Fujimoto disease, and Castleman disease. Kikuchi–Fujimoto disease typically shows histiocytic necrotizing lymphadenitis with prominent reactive pDCs but lacks clonal markers. SLE-associated pDC proliferation is similarly reactive and nonclonal. 4
Distinguishing MPDCP from these entities requires integrated assessment of histology, immunophenotype, clinical context, and molecular studies.
Therapeutic Implications
While MPDCP may not require targeted treatment, its presence may be an indicator of disease progression in CMML. Hypomethylating agents remain the core treatment for CMML.
Novel therapies targeting CD123 which is overexpressed in pDCs and CMML cells offer potential for future treatment strategies. Tagraxofusp, a CD123-directed toxin, is approved for BPDCN and is under investigation for broader use. Vibecotamab, a CD3-CD123 bispecific antibody, has shown activity in high-risk myeloid neoplasms. Ruxolitinib, a JAK1/2 inhibitor, demonstrated symptom and spleen size reduction in large proportion of CMML patients in a phase II trial. 27 These findings suggest JAK-STAT pathway inhibition may benefit CMML-related MPDCP.
Further research is needed to assess the efficacy of emerging therapies for CMML and associated MPDCP. This research is warranted to evaluate these agents’ efficacy in targeting clonal pDCs without impairing host immune function.
Conclusion
This case underscores the diagnostic complexity of MPDCP, particularly within myeloid neoplasms like CMML. Accurate diagnosis necessitates a comprehensive evaluation to differentiate MPDCP from entities such as BPDCN, given their overlapping immunophenotypic features. Notably, while elevated Ki-67 and TdT expression broaden the spectrum of MPDCP, the absence of CD56 expression remains a distinguishing factor. The persistence of ASXL1 and CBL mutations further links MPDCP to CMML, highlighting the need for vigilant monitoring for potential progression to AML.
Future research should focus on exploring CD123-directed therapies and elucidating the role of pDCs within the microenvironment of myeloid neoplasms. Genomic and transcriptomic analyses are essential to deepen our understanding of MPDCP’s pathogenesis and its clinical implications. Incorporating MPDCP into the differential diagnosis of CMML-related lymphadenopathy and ensuring long-term patient monitoring are critical. As our molecular insights advance, targeted therapeutic strategies may emerge, offering improved management for these rare and complex cases.
Footnotes
Acknowledgements
Not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Verbal informed consent was obtained from the patient for their anonymized information to be published in this article.