
Abstract
Chronic lymphocytic leukemia (CLL) is an indolent malignancy characterized by the accumulation of dysfunctional B-cell lymphocytes. Complications such as hemophagocytic lymphohistiocytosis (HLH) can arise, particularly during disease progression. HLH has been increasingly reported as a complication of CLL, often triggered by factors such as superimposed infections, chemotherapy, Richter transformation, or disease progression. This case explores HLH as an initial presentation of undiagnosed CLL without any identifiable trigger. We present the case of a 65-year-old woman who presented with a high-grade fever, sore throat, and pancytopenia. Despite broad-spectrum antibiotic treatment, her condition deteriorated. Investigations revealed elevated ferritin levels, low natural killer cell activity, and other findings consistent with HLH. Flow cytometry and bone marrow biopsy ultimately confirmed the diagnosis of CLL. HLH is characterized by the hyperactivation of immune cells and is known to be triggered by a variety of factors, including infections and malignancies. In this case, the absence of identifiable triggers raises important questions about the underlying pathophysiology linking HLH with CLL. While previous reports have highlighted HLH as a complication of CLL, typically secondary to infection or treatment, this case is particularly noteworthy due to the unexplained onset of HLH in the absence of such triggers. This case underscores the need for heightened awareness of HLH as a potential manifestation of underlying malignancy, especially in non-septic patients presenting with unexplained fever and pancytopenia. In addition, the simultaneous presentation of normal pressure hydrocephalus emphasizes the complex interplay of inflammatory processes in CLL. Further research is needed to explore the relationship between inflammation and the pathogenesis of CLL.
Introduction
Chronic lymphocytic leukemia (CLL), one of the 4 major subtypes of leukemia, is an indolent lymphoproliferative disease resulting from the proliferation and accumulation of morphologically mature but immunologically dysfunctional B-cell lymphocytes. It is the most prevalent adult leukemia in the United States, accounting for 25% to 30% of all leukemias.1,2
Chronic lymphocytic leukemia is characterized by the clonal expansion of CD5+CD23+ B cells in the blood, marrow, and secondary lymphoid tissues. This condition may arise from insults to the B-cell clone caused by genetic mutations or changes in the bone marrow and lymph node environment. As a result, functionally incompetent lymphocytes accumulate, leading to lymphadenopathy, hepatosplenomegaly, and cytopenias. 1
Chronic lymphocytic leukemia is commonly found in asymptomatic patients who have incidental lymphocytosis noted on routine blood tests performed for other reasons. 3 A small fraction of patients present with B symptoms, hepatosplenomegaly, lymphadenopathy, or skin lesions (leukemia cutis). Other sequelae of CLL, such as anemia and thrombocytopenia from hypercellular bone marrow and splenomegaly, are not uncommon initial manifestations in some patients. The lack of functional B cells in CLL leads to hypogammaglobulinemia, which may result in recurrent infections.4-6 Infrequently, patients with CLL present with acute aggressive lymphadenopathy, which could result from Richter transformation into aggressive diffuse large B-cell lymphoma and, more rarely, Hodgkin lymphoma or T-cell lymphomas. 4
The association and increased incidence of hemophagocytic lymphohistiocytosis (HLH) in CLL are rarely reported in the literature. HLH is found to occur in untreated, progressive, and relapsed CLL. It is usually triggered by infections, disease progression, treatment initiation, or as a consequence of targeted treatment for CLL. 7 This is explained by an aberrant and robust immune response to the trigger or cytotoxic agent. However, the spontaneous occurrence of HLH as the initial presentation of previously undiagnosed CLL has never been reported. While cases of normal pressure hydrocephalus (NPH) associated with HLH are present in the literature, the simultaneous occurrence of NPH and HLH in this setting warrants detailed exploration, which we aim to address in this study.
Case Description
We report a 65-year-old woman with a history of Chiari type 1 malformation who presented with high-grade fever, sore throat, coryza, headache, and presyncope. Despite taking amoxicillin-clavulanic acid for 7 days, her symptoms persisted. On admission, her vitals showed a temperature of 104.2°F, heart rate of 137, and blood pressure of 95/61. Physical examination was largely unremarkable, and neurological assessment showed no deficits. Laboratory results revealed pancytopenia (Hb 8.8, white blood cell [WBC] count 1000, absolute neutrophil count [ANC] 0, platelets 58 000). Computed tomography (CT) of the chest, abdomen, and pelvis with contrast showed hepatomegaly with the right lobe of the liver measuring 19 cm, multiple enlarged mediastinal lymph nodes, and no evidence of splenomegaly. CT of the head suggested NPH.
Consultation with hematology-oncology led to a differential workup for pancytopenia, including potential bone marrow infiltration and aplasia (Table 1). Labs were sent, and the patient was placed under strict reverse isolation due to neutropenia. Medication reconciliation was done, and the patient was not found to have taken any drugs causing pancytopenia. Given the persistent fever and negative autoimmune workup, broad-spectrum antibiotics were started, but the patient remained febrile. HLH was suspected, and subsequent tests indicated significant findings: low natural killer (NK) cell count and high ferritin. The patient met 5 out of 8 HLH criteria per the HLH 2004 diagnostic guidelines:
• Fever of 104.3°F,
• Peripheral blood cytopenia in 2 cell lines (WBC 0.8; ANC 0.0; Hb 10.0; Platelets 66 000),
• High ferritin level of 8587,
• Low NK cells (CD3-CD16+CD56+ (abs) = 41; CD3-CD16+CD56+ (%) = 9%),
• Low CD25 of 6482.
Results of Pancytopenia Workup.

A diagnosis of HLH was confirmed, and treatment with etoposide and dexamethasone was initiated. The patient improved dramatically, becoming afebrile within 24 hours. Antimicrobials were planned to be continued until the ANC exceeded 500, and no fever spikes occurred for more than 48 hours after reaching the target ANC. Flow cytometry revealed a small monoclonal B-cell population indicative of CLL, which was confirmed by bone marrow biopsy (Figures 1-6). She was referred to a tertiary center for further management of HLH and chemotherapy for CLL, while neurology recommended supportive care for NPH.

Bone marrow biopsy—CD3 staining.
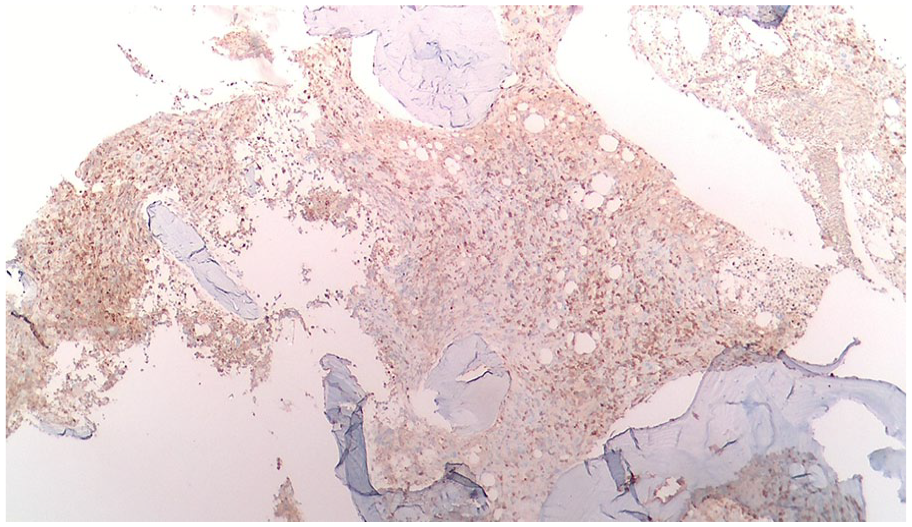
Bone marrow biopsy—CD5 staining.

Bone marrow biopsy—CD20 staining.
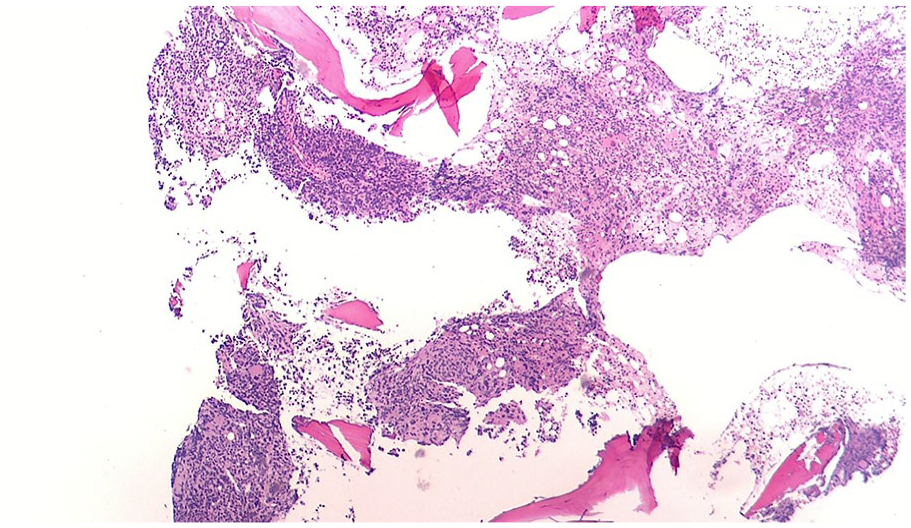
Bone marrow biopsy—H&E 40×.

Bone marrow biopsy—H&E 100×.

Bone marrow biopsy—H&E 200×.
Discussion
HLH is a hyperinflammatory condition characterized by dysregulated activation of NK cells, CD8+ T lymphocytes, and macrophages.7 According to the HLH-2004 Diagnostic Criteria, HLH can be diagnosed through either A or B:
A. A molecular diagnosis with an HLH-associated genetic mutation (homozygous or compound heterozygous in children; heterozygous in adults if clinical presentation is consistent).
B. Any 5 of the following 8 clinical and laboratory criteria: fever >38.5°C, splenomegaly, cytopenia (affecting ≥2 of 3 lineages), hypertriglyceridemia/hypofibrinogenemia, hemophagocytosis in tissues, low or absent NK cell activity, serum ferritin ≥500 μg/L, or soluble CD25 ≥2400 U/mL.
HLH can be primary or secondary, with secondary HLH most commonly triggered by infections, malignancies, autoimmune diseases, and metabolic disorders. The H Score, 8 which assesses HLH probability, indicated a score of 186 in our patient, suggesting a 70% to 80% likelihood of hemophagocytic syndrome.
Various differentials were considered and ruled out, including sepsis (no infectious foci), autoimmune disease (negative anti-nuclear antibody and rheumatoid factor), drug reactions (normal eosinophils, no rash), posterior reversible encephalopathy syndrome (no neurological symptoms), and absence of preexisting connective tissue diseases.
These findings, along with meeting the 2004 HLH criteria, confirmed HLH as the cause of symptoms in the context of newly diagnosed CLL. HLH associated with malignancies often has a higher mortality and can be misdiagnosed due to symptom overlap with other conditions.9,10 Multiple case reports of HLH in CLL have been published, all falling under 3 domains: HLH triggered by infections, chemotherapy, or changes in disease course.
Rare occurrences of HLH in CLL with Histoplasmosis have been described, where subsequent investigations, including autopsy, revealed no obvious triggers for HLH other than histoplasma infection.11,12 During the pandemic, a case of HLH in CLL triggered by H1N1 (Influenza A) infection was also reported. 13 Epstein-Barr virus infection, associated with the precipitation of HLH in CLL, is also theoretically possible.14,15
While indolent CLL is historically treated with supportive measures, patients who meet the criteria for treatment are generally managed with immunotherapy, radiation, and chemotherapy. HLH is reported as a complication of checkpoint inhibitor therapy for CLL.16 A significant number of HLH cases have been documented as a result of initiating CLL treatment with ibrutinib.14-17 This association was strongly supported by a Johns Hopkins case series of 3 cases of CLL-associated HLH manifesting after ibrutinib initiation. 18 Presentation of HLH after chemotherapy with rituximab, cyclophosphamide, bendamustine, and fludarabine has also been studied, which could be attributed to B-cell suppression by rituximab or T-cell suppression by fludarabine.12,19-21
The diagnosis of HLH without evidence of apparent infections or exposure to cytotoxic agents has also been reported. In such cases, untreated CLL was the only identified trigger. 22 Spontaneous emergence of HLH in treatment-resistant CLL, 23 progression of CLL,24,25 persistent CLL, 26 and relapse of CLL14,17,27 have also been described. A combined influence of infectious triggers, cytotoxic agents, and/or disease transformation also leads to HLH in a handful of CLL patients.14,15,17
HLH presents significant diagnostic and therapeutic challenges due to its nonspecific symptoms and variety of triggers. It is rarely reported in the context of CLL, typically associated with infection, chemotherapy, or disease transformation. Our case highlights a unique instance where HLH was the initial manifestation of CLL without these classic triggers, marking the first such report. This underscores the importance of ruling out indolent malignancies like CLL in patients with HLH.
Emerging studies suggest that inflammation may play a role in NPH, 28 as seen in our patient with Chiari type 1 malformation. The presence of NPH may indicate widespread inflammation, challenging the perception of CLL as purely indolent. This raises questions about the relationship between early inflammatory processes and CLL’s pathogenesis, warranting further research into potential molecular therapies targeting inflammation in CLL.
Overall, this case emphasizes the need for a high index of suspicion in non-septic patients with fever and unexplained pancytopenia, as timely interventions in HLH are critical for prognosis. In addition, the unexplored hereditary links between CLL and HLH present an intriguing area for future investigation.
Conclusion
In conclusion, this case highlights the rare and unexplained presentation of HLH as the initial manifestation of undiagnosed CLL, occurring without the typical triggers such as infection or chemotherapy. It emphasizes the importance of considering underlying malignancies like CLL in the differential diagnosis of HLH, particularly in non-septic patients with unexplained fever and pancytopenia. Further research into the relationship between inflammation, HLH, and CLL pathogenesis is essential to enhance our understanding and improve treatment strategies for these complex interactions.
Footnotes
Acknowledgements
We would like to express our gratitude to the clinical staff at The Brooklyn Hospital Centre for their exceptional care and support during the management of the patient described in this case report. We also thank our colleagues for their insightful discussions and contributions to the development of this manuscript. Finally, we appreciate the guidance and resources provided by the Institutional Review Board, which facilitated the ethical approval for publication.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Permission to report this case was granted by the Institutional Review Board of The Brooklyn Hospital center, Number: 2263174.
Informed Consent
Written informed consent was obtained from the patient for their anonymized information to be published in this article.