
Abstract
Rafiq syndrome, MAN1B1-CDG, was described in 2010 and associated with genetic mutation in MAN1B1 gene in 2011. The disorder follows an autosomal recessive pattern of inheritance and typically presents with specific facial dysmorphism, intellectual disability, developmental delay, obesity, and hypotonia. The syndrome belongs to a group of metabolic disorders called Congenital Glycosylation Disorders (CGD). In this study, we discuss a 5-year-old male from Palestine who presented with developmental delay, hypotonia, characteristic facial dysmorphisms, impulsive behaviors, inability to speak, cryptorchidism, and other manifestations. This constellation of manifestations raised suspicion of a genetic disorder, prompting whole exome sequencing (WES), which revealed the presence of a homozygous likely pathogenic variant in the MAN1B1 gene (c.1976T>G)(p.Phe659Cys). We also reviewed all previously documented cases and compared the clinical features among them. After reviewing the family pedigree and its suspected cases, we found that the 2 most frequent features among them are intellectual disability and facial dysmorphism, whereas the least frequent one is truncal obesity. We discussed the importance of providing genetic counseling to parents of children with this and other rare, autosomal recessive disorders to prevent new cases from appearing.
Introduction
Rafiq syndrome, MAN1B1-CDG, an identified genetic condition, was documented for the first time by Dr Rafiq and his colleagues in 2011. 1 In their study, Dr Rafiq and his team observed a significant association between intellectual disabilities, developmental delay, and specific facial dysmorphism in 12 cases in 5 families from Pakistan and Iran. Genetic studies of those 12 cases revealed 3 (likely or known pathogenic) variants in the MAN1B1 gene in all cases, causing enzyme deficiencies leading to a Type II Congenital Glycosylation Disorders (CGDs), 2 which is now recognized as the underlying cause of Rafiq syndrome.
Congenital Glycosylation Disorders are a group of rare autosomal recessive metabolic disorders that result from a defect in the N-glycosylation process, which constitutes approximately 90% of the body’s glycosylation processes.3,4 The defect can manifest either during the assembly process in the cytosol and endoplasmic reticulum (CGD type I) or in the Golgi apparatus (CGD type II). 4
Subsequent studies have reported an additional 30 cases of Rafiq syndrome in 20 families.2,5 Various MAN1B1 gene mutation variants have been documented, including missense, frame-shift, and splice mutations, with some presenting as homozygous and others as heterozygous. In this study, we present a case of Rafiq syndrome from Palestine involving a previously documented MAN1B1 gene mutation. In addition, we reviewed the patient’s family pedigree, noting several family members exhibiting clinical manifestations/phenotype suggestive of Rafiq syndrome who did not undergo genetic testing (suspected cases). Furthermore, we conducted a comprehensive review of all previously reported cases to enhance our understanding of this syndrome.
Methods
We searched in Scopus, Pubmed/NIH, Embase, OMIM, and Hand search from 2011 until 2022 using the following keywords: “MAN1B1 mutation,” “Rafiq Syndrome,” and “Case Report,” and we included every single case when the complete text was done.
We also use SPSS to analyze the data we gathered to point some common associations between cases.
Case Presentation
In this study, we present a 5-year-old male child from Palestine who was referred to the genetic clinic due to dysmorphism, hypotonia, bilateral ptosis, and behavioral abnormalities. He is the second child of healthy consanguineous Palestinian parents—first cousins once removed; he was born at term, and his weight and length were 3.7 kg and 52 cm, respectively; the perinatal period was uneventful, with no complications.
Since infancy, he was noticed to have mild developmental delay starting with suboptimal sucking at birth, delayed fine and gross motor development (manifesting as: sitting without support at 9 months, walking at 21 months, and going up and down stairs at 26 months). Speech delay was noted, characterized by an absence of expressive language despite adequate receptive language skills.
He was diagnosed with cow’s milk protein allergy and severe dermatitis at age of 2.5 years and with cryptorchidism, which warranted a bilateral orchiopexy.
At this age (5 years), his speech was largely incomprehensible, with only a few words used. However, he could understand spoken language and interact with family members and peers. He remained untrained in using the toilet, was often uncooperative and easily distracted, and exhibited aggressive behavior. Clinical examination revealed notable dysmorphic features, such as a high broad forehead, a short chin (Figure 1), hypertelorism, epicanthal folds, bilateral ptosis, right eye squint (Figure 2), full lower lip, large mouth, wide-spaced teeth (Figure 3), high-arched palate, and a large protruding geographic tongue. His skin appeared dry and pale, and ulcers were present on both hands (Figure 4A); he had tapering fingers, relatively moderate bilateral 2 to 3 syndactyly of toes of both feet and flat feet (Figure 4B). He had severe dental caries and drooling. His weight and height were more than 50th and 90th percentiles, respectively. Otherwise, he showed no signs of truncal obesity, overeating behavior, chest deformity, joint hypermobility, or scoliosis. Examination of other systems was free.

(A and B) Anterior and lateral view of the patient head shows a high broad forehead, a short chin, and neck.

Eyes show hypertelorism, epicanthal folds, bilateral ptosis, and right eye squint.

Full lower lip, large mouth, and wide-spaced teeth.

(A) An erythematous flat ulcer (red arrow) and tapering fingers and (B) flat feet and relatively moderate bilateral 2 to 3 syndactyly of toes of both feet.
His clinical features were suggestive of a genetic/syndromic diagnosis, so genetic work up was performed which confirmed the diagnosis of Rafiq syndrome (see discussion). Other metabolic tests were ordered and were normal.
Family History and Pedigree Analysis
Rafiq syndrome shows an autosomal recessive pattern of inheritance, and some of our case relevant shows similar clinical features as intellectual disability (ID) and facial dysmorphism, so we conducted Family pedigree for 3 generations to point abnormal cases (Figure 5) and subsequently to compare them to our case (Table 1).

The family pedigree for 3 generations, and the suspected cases highlighted with blue light.
The relatives who have similar features to our case (Rafiq syndrome).
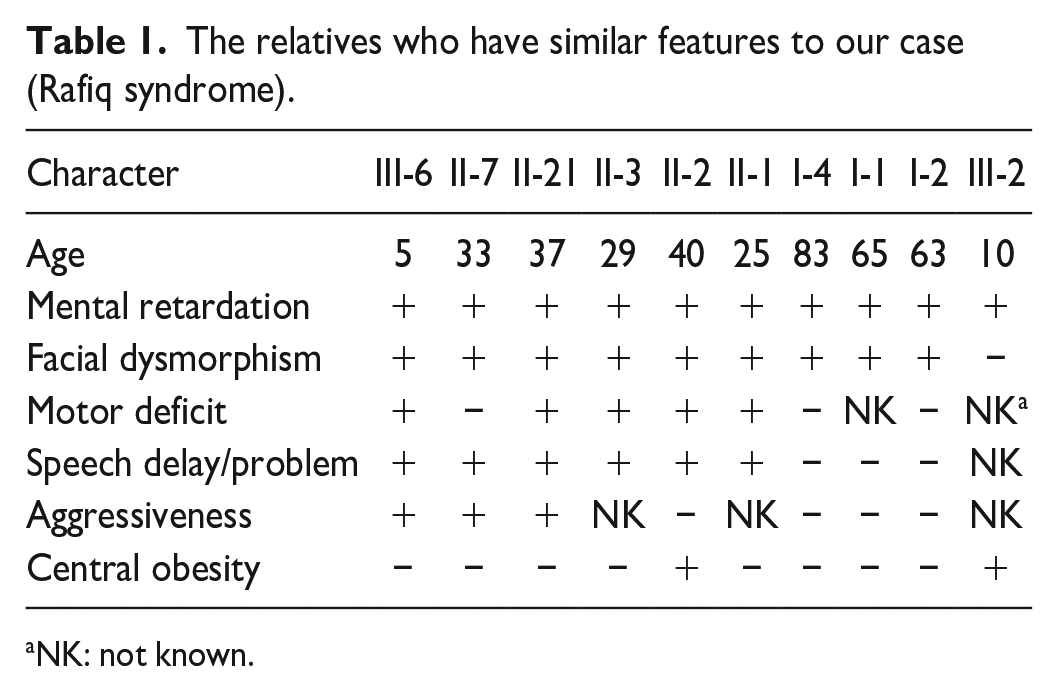
NK: not known.
Upon reviewing the extended family data, our case is the only family member diagnosed through genetic testing. However, other suspected cases exhibiting clinical symptoms or phenotypes of Rafiq syndrome have not undergone genetic testing, as shown in Table 1.
The patient’s uncle (II-7), a 33-year-old man, is the closest suspected case, displaying all the characteristic features observed in our case, including mental retardation, facial dysmorphism, speech difficulties, and aggressiveness. The only notable difference is the absence of motor delay. In addition, he has cryptorchidism, which may be related to the syndrome or could be an isolated finding.
The family has a strong cultural tendency to marry within close relatives, as evidenced by the frequent consanguinity observed across generations in the pedigree. This high frequency of consanguinity increases the risk of multiple individuals carrying the same mutation, resulting in a higher likelihood of homozygous individuals manifesting the disease due to the autosomal recessive nature of Rafiq syndrome.
Regarding the phenotypes of suspected cases, we observed that the characteristic features (phenotype) tend to be more pronounced, severe, and present at an earlier age as we progress through generations.
In the table of suspected cases, all exhibited ID and facial dysmorphism, except for case (III-2). These 2 features were selected as criteria for identifying suspected cases. The least common feature among all cases was truncal obesity, which was also absent in our case.
Literature Review
Going back to the previous published research, a review of 44 cases of Rafiq syndrome including our case was done.
We compared Nemours characteristics like ID, consanguinity, motor developmental delay, and dysmorphic features in Table 2.
A literature review of 44 cases of Rafiq syndrome including our case, which compared the reported mutations and the clinical features.
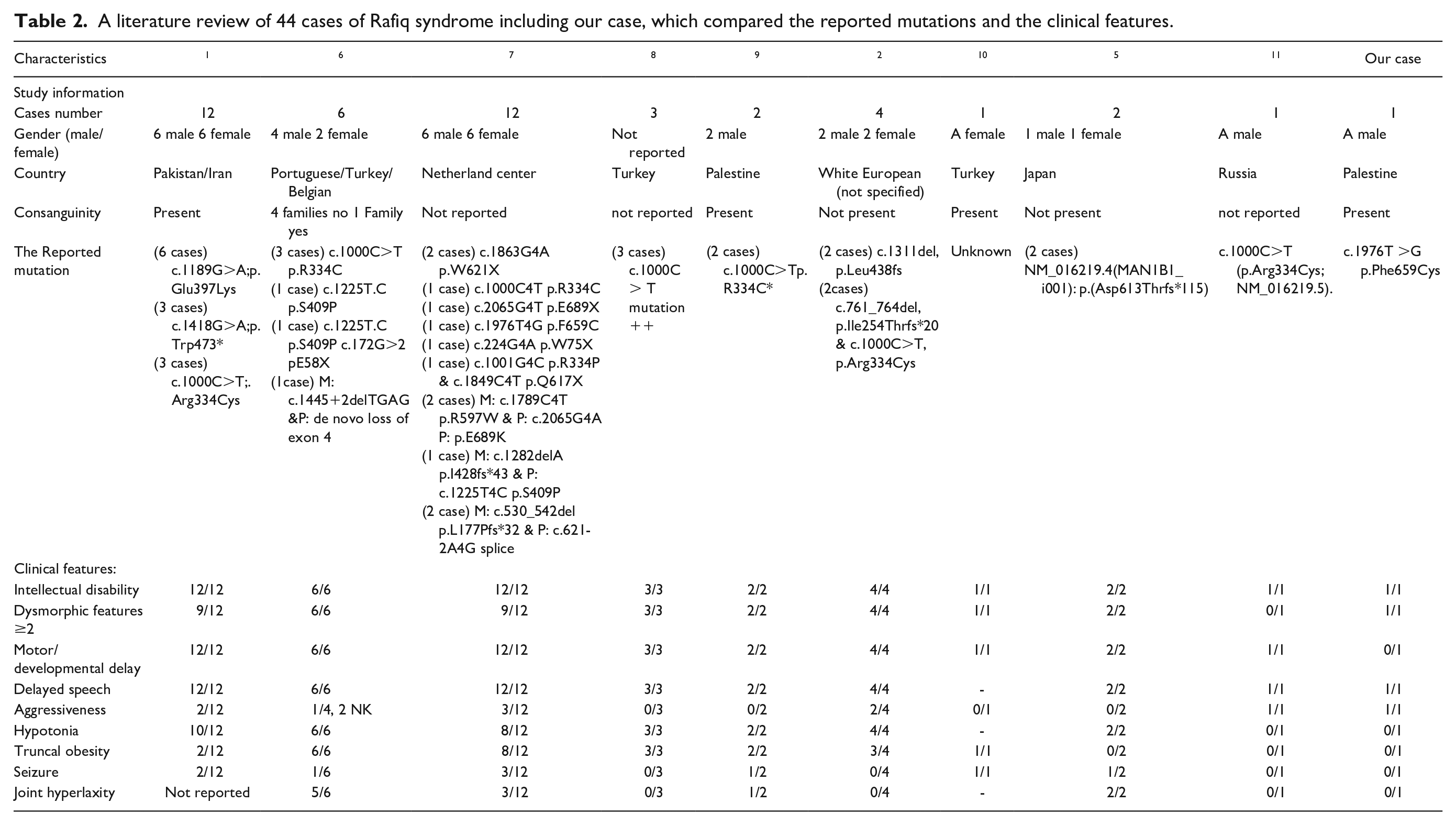
A review of 44 cases of Rafiq syndrome, including our own, was conducted based on previously published research. As shown in Table 2, 10 studies have been published that compare factors such as consanguinity, male/female distribution, developmental abnormalities, seizures, and joint hyperlaxity. However, upon reviewing these studies, we identified missing information that could further contribute to a better understanding of this condition.
Statistical analysis reveals that the c.1189G>A; p.Glu397Lys mutation is the most frequently observed mutation. Seizures were reported in 16% of cases, whereas 34% of cases presented with aggressive behavior. Regarding ID, the distribution was as follows: 48% of cases had mild ID, 30% had moderate, and 11% had severe ID, with 1 case lacking IQ data. Finally, dysmorphic features were observed in 84% of cases, with at least 2 or more dysmorphic traits present.
Discussion
In this case, we describe a 5-year-old male from Palestine who was diagnosed with Rafiq syndrome as the pediatricians observed some overlapping features with Coffin-Lowry syndrome and Smith-Lemli-Opitz syndrome, so whole exome sequencing (WES) was done and revealed the presence of a homozygous likely pathogenic variant in the MAN1B1 gene (c.1976T>G)(p.Phe659Cys).
As ID may have a genetic basis, specific symptoms and dysmorphisms can aid the genetic diagnosis. 12 Homozygous pathogenic variant in this gene is associated with Rafiq syndrome which is an autosomal recessive disorder characterized by variably impaired intellectual and motor development, a variable facial dysmorphism, truncal obesity, and hypotonia. Behavioral problems, including overeating, verbal and physical aggression, have been reported in some cases.
Although other chronic granulomatous diseases (CDGs) can manifest as failure to thrive, frequently due to feeding issues and/or protein-losing enteropathy, it is noteworthy that nearly all documented instances of Rafiq syndrome, MAN1B1-CDG, exhibited normal growth by length and weight gain consistent with their age and gender expectations.13,14 One possible explanation could be that the specific pathogenic variant in this gene may affect glycosylation pathways that are less directly involved in growth regulation. In addition, there might be compensatory mechanisms that allow for normal growth despite the presence of the variant; so we need further investigations into the specific functions of this gene and its role in growth regulation, which could provide more clarity on this phenomenon.
The identified variant in our case has been reported before in a female patient with overlapping clinical phenotype. According to the study of that case, a recognizable profile in routine CDG screening by transferrin isoelectric focusing with an isolated increase of trisialotransferrin was confirmed in that patient, as well as other patients included in the study.
Back to our case, the patient had mild mental and motor disability along with aggressive behavior; otherwise, he had no other symptoms of MAN1B1 features such as joint hyperlaxity, truncal obesity, and dolichocephaly.
It is worth mentioning that our patient was also diagnosed with cow’s milk protein allergy, severe dermatitis, and cryptorchidism; these features could be isolated findings or could be related to his, although it has not been previously documented in Rafiq syndrome cases. This raises important questions about potential shared pathophysiological mechanisms among different CDGs. Although the previous hypotheses can still be applied—whether as an isolated finding or as part of the syndrome—we can also note that there may be a hereditary factor involved, as his uncle also had this condition.
Recommendations for the Family
We discussed with parents that this condition is inherited in an autosomal recessive manner; so each pregnancy of a couple who have had a child with Rafiq syndrome has an approximately 25% chance of producing an affected child, an approximately 50% chance of producing an asymptomatic carrier, and an approximately 25% chance of producing an unaffected child who is not a carrier.
We recommend them to arrange parental testing to confirm the carrier status of both parents for the identified variant in MAN1B1 gene (c.1976T>G)(p.Phe659Cys). As well as for his 2 sisters (a 5-year-old healthy child and a 1-month-old who is apparently healthy till the moment) and for his uncle (who is suspected to be affected).
For further validation of the variant’s pathogenicity, we recommended performing diagnostic genetic screening using transferrin isoelectric focusing. We discussed the couple’s options for future pregnancies, including invasive prenatal genetic testing (Chorionic Villus Sampling vs amniocentesis) and in vitro fertilization with preimplantation genetic diagnosis (IVF/PGD).
Conclusion
We present the first reported case of Rafiq syndrome in a Palestinian patient, with genetic testing confirming the presence of an MAN1B1 gene mutation. This mutation was previously described only in a single female patient exhibiting clinical features suggestive of Rafiq syndrome. In this report, we describe the clinical presentation of this patient, along with 9 other family members carrying the same mutation, thereby expanding the understanding of the clinical and molecular aspects of the disease. The patient exhibited typical dysmorphism, as well as mild mental and motor disabilities, in addition to painful defecation, severe dermatitis, cryptorchidism, and aggressive behavior. Notably, he did not display other common features of MAN1B1 mutations, such as joint hyperlaxity, truncal obesity, or dolichocephaly.
Counseling for the Family
We recommended that the family undergo genetic testing for both previously diagnosed and any new suspected cases to enable early diagnosis. In addition, we advised them to avoid consanguineous marriages to reduce the risk of perpetuating the mutation. We also encouraged them to minimize marriage between relatives as much as possible to decrease the degree of consanguinity, thereby reducing the likelihood of new cases and the risk of other autosomal recessive disorders, including those beyond Rafiq syndrome.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Verbal informed consent was obtained from the parents of the patient for their anonymized information to be published in this article.