
Abstract
Pierpont syndrome is a rare and recently described multiple congenital anomaly syndrome, classically characterized by global developmental delay, distinctive facial dysmorphic features, and abnormal fat distribution in distal limbs. Only few cases were previously documented. We report a case of a term male neonate admitted to the neonatal intensive care unit because of feeding difficulties. Intrauterine growth restriction, microcephaly, and bilateral equinovarus foot were diagnosed in the second trimester, and prenatal array comparative genomic hybridization showed no abnormality. Physical examination revealed bilateral flexion deformities of wrists, elbows, knees and clubfoot, large hands and feet, deep palmar and plantar grooves, and calcaneo-plantar fat pads. Craniofacial dysmorphism, axial hypotonia, and hypoactivity were also observed. Due to the presence of congenital and non-progressive joint contractures, arthrogryposis multiplex congenita (AMC) was considered. A comprehensive diagnostic workup, including a Next Generation Sequencing target panel, was performed but did not establish a diagnosis. The clinical exome identified an heterozygous pathogenic variant in the TBL1XR1 gene (NM_001321194.1: c.1337A>G, p.[Tyr446Cys]), allowing Pierpont syndrome diagnosis. Our case stands out for reporting the novel AMC presentation in a Pierpont syndrome newborn. The broader and precocious genetic testing proved to be an essential clarifying diagnostic tool. Our patient supports the relation between the p.Tyr446Cys sequence variant in TBL1XR1 gene with this rare syndrome, reinforcing its association with a distinctive and recognizable phenotype, as well as expanding its clinical features to include AMC.
Keywords
Introduction
Pierpont syndrome (OMIM #602342) is a rare multiple congenital anomaly syndrome, classically characterized by global developmental delay, distinctive facial dysmorphic features, and abnormal fat distribution in distal limbs.1,2 Until now, few cases have been reported, and the most recent ones have documented new additional associated features.3-6 A mutation in TBL1XR1 gene on chromosome 3q26 was only recently identified as being associated with the syndrome, which may contribute to its clinical spectrum’s broadness still not being totally clarified. 7
Arthrogryposis multiplex congenital (AMC) is characterized by the presence of congenital and non-progressive joint contractures affecting at least 2 different body areas.8-11 Involved joints become permanently fixed in a flexed or extended position, totally or partially limiting its movement. 12 Distal joints are more frequently and severely affected than proximal, with clubfoot and flexion deformities of wrists being the most common manifestations. 13 Its incidence is of approximately 1/3000 to 1/5000 live births, and its clinical severity can range from an isolated joint contracture to lethal disorders.9,12,14,15
AMC constitutes a descriptive presenting term rather than a specific diagnosis.9,11,12 It has been described in more than 400 clinically and genetically heterogeneous disorders, representing a diagnostic challenge.11,16 To date, chromosomal abnormalities and more than 320 single gene disorders were already associated, with new causes being identified regularly.11,17 However, many cases remain of an unknown etiology. Therefore, genetic testing has progressively gained a crucial role in AMC patients’ investigation.
Herein, we report a case of a newborn presenting with notorious AMC, in whom clinical exome sequencing unveiled a Pierpont syndrome diagnosis. To our knowledge, this is the first report of arthrogryposis in this uncommon syndrome, in whom broader genetic investigation was an essential tool for an accurate diagnosis.
Case Report
A term male neonate was born at 41 weeks and 1 day by vacuum-assisted delivery. His Appearance, Pulse, Grimace, Activity and Respiration (APGAR) score was 7/9/10 and resuscitation measures were not needed. His birth weight, length, and head circumference were 3145 g, 51 cm, and 31.5 cm (3rd-10th, 10th-50th, and <3rd percentile according to Fenton growth charts, respectively). Pregnancy was properly monitored and intrauterine growth restriction, microcephaly (<3rd percentile intrauterine growth), and bilateral equinovarus foot were diagnosed in second trimester ultrasound. An amniocentesis was performed at 20 gestational weeks and revealed a 46, XY karyotype. Prenatal array comparative genomic hybridization (aCGH) analysis showed no abnormality. He was a first child of non-consanguineous healthy parents, and family history was nonrelevant.
During his first hour of life, he was admitted to the neonatal intensive care unit (NICU) because of feeding difficulties and hypoglycemia. At admission, he presented axial hypotonia, hypoactivity, and weak sucking reflex. His examination also revealed some peculiar physical features, including distinctive craniofacial dysmorphic characteristics such as broad nasal bridge and tip with anteverted nostrils, thin upper lip vermillion, right preauricular sinus and auricular lobe indentation, flat occiput, and bifid uvula (Figure 1A and D). Widely spaced nipples and bilateral cryptorchidism were also observed. However, the most notorious aspects of the physical examination were prominent bilateral flexion deformities of wrists, elbows, knees, and clubfoot, with apparent large hands and feet with deep palmar and plantar grooves, and calcaneo-plantar fat pads (Figure 1B-D).
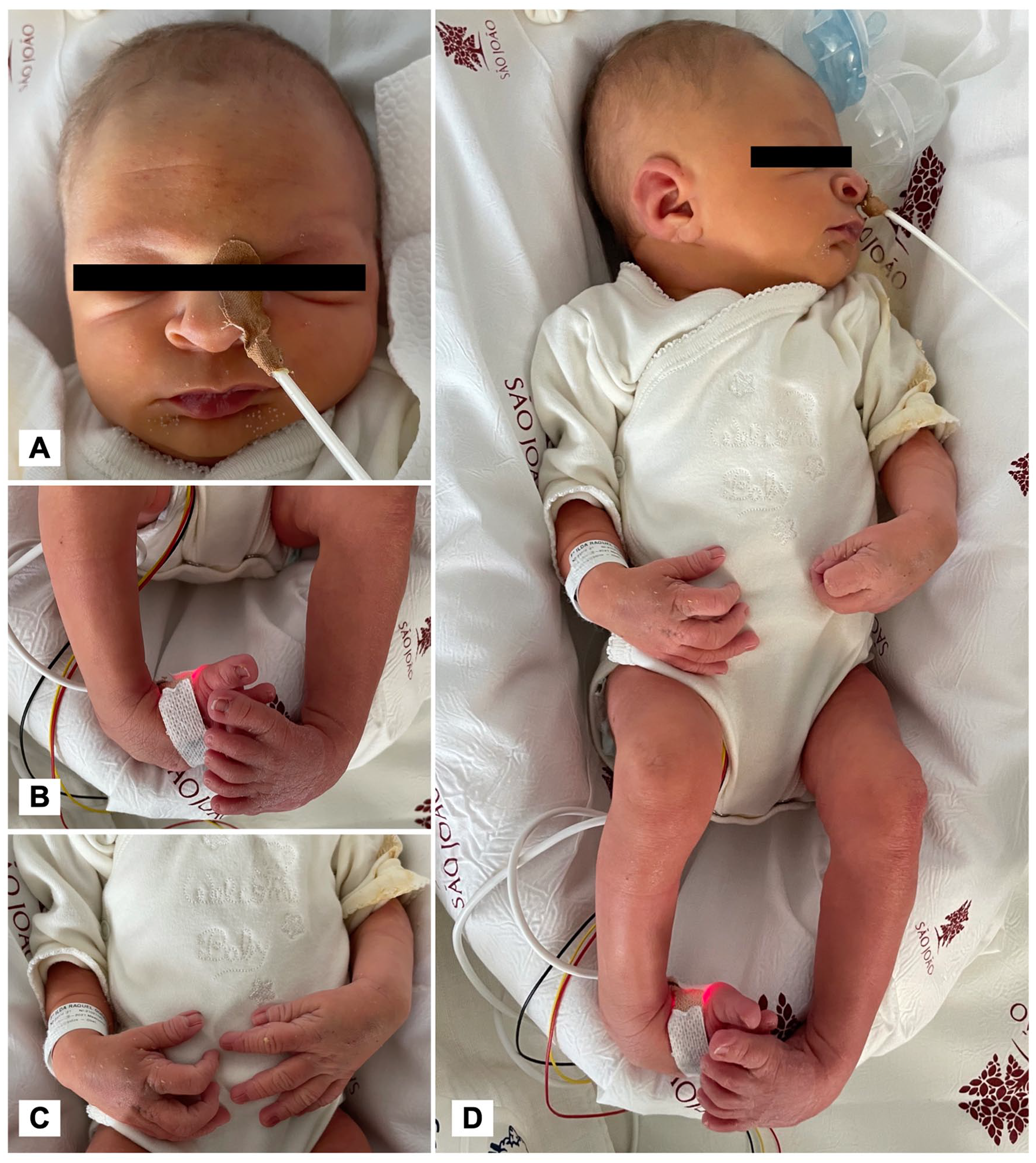
Patient’s distinctive physical examination features: (A) Distinctive craniofacial features, including deep nasal bridge, broad nasal tip with anteverted nostrils, and thin upper lip vermillion. (B) Bilateral equinovarus foot. (C) Flexion posture of wrists and elbows and large hands, with deep grooves. (D) Right preauricular sinus and auricular lobe indentation, flat occiput, and flexion posture of joints above described.
Due to the presence of congenital and non-progressive joint contractures, AMC was considered and a comprehensive diagnostic workup performed. Whole body radiography only confirmed the presence of bilateral clubfoot. Abdominal and renopelvic ultrasonography revealed a right duplex kidney. Ophtalmologic examination, otoacoustic emissions testing, serum creatine kinase, electromyography, and brain magnetic resonance imaging (MRI) were normal. Medular MRI pointed an incipient left convexity scoliosis due to an inversion of T8 and L2 vertebral bodies configuration. As previous investigation did not display an accurate diagnosis, and a syndromic situation was suspected, precocious genetic testing was performed. A Next Generation Sequencing (NGS) target panel of 87 genes associated with syndromic/nonsyndromic AMC did not find any potentially pathogenic variant. The clinical exome sequencing (CES) identified a de novo heterozygous pathogenic variant in the TBL1XR1 gene (NM_001321194.1: c.1337A>G,p.[Tyr446Cys]), establishing the Pierpont syndrome diagnosis. The patient remained in the NICU for 35 days due to prolonged feeding difficulties, requiring gastric tube feeding support. Multidisciplinar follow-up was guaranteed, including local physical, developmental, and palliative care team support.
At 12 months of age, he presented global developmental delay with significant hypotonia, not being able to sit unsupported. He was capable of soft foods’ oral feeding but was incapable of eating solid ones. His hearing was normal, and he babbled. A right eye small subcapsular cataract was diagnosed at 11 months old and he had orchidopexy for bilateral cryptorchidism. Bilateral equinovarus foot was successfully treated by Ponseti method, and the remaining joint contractures improved after physiotherapy, without any movement limitation still being observed. Global classic physical features of Pierpont syndrome were well noted at this age (Figure 2). Distinctive craniofacial features were essentially unchanged, but became more pronounced than during neonatal period, including broad face with midface hypoplasia, high forehead, high anterior hairline, deep-set eyes with narrowed palpebral, broad nasal bridge and tip with anteverted nostrils, and thin upper lip vermillion (Figure 2C). Digital typical findings were also still noticeable, including abnormal fat distribution in the hands and feet, as well as typical fetal fingers and deep palmar and plantar grooves (Figure 2A and B).

Twelve-month-old patient images showing characteristic features of Pierpont syndrome: (A) Corrected equinovarus foot and abnormal foot’s fat distribution. (B), Typical deep palmar grooves and fetal fingers. (C), Distinctive craniofacial features, including a broad face, midface hypoplasia, high forehead, high anterior hairline, deep-set eyes with narrowed palpebral, broad nasal bridge and tip with anteverted nostrils, long smooth philtrum, and thin upper lip vermillion.
Discussion
Pierpont syndrome was first described in 1998, when Pierpont et al 1 documented 2 patients with plantar fat pads, characteristic facial aspects, and developmental delay. 3 Since then, it has been documented as a rare disorder with distinct features from 4 main areas, which are common across most of the patients, including the one we report: craniofacial features, findings in the hands and feet, neurodevelopment disorders, and feeding and growth problems. 3
To date, less than 15 cases were reported, and its etiology was unknown until recently, when Heinen et al 7 identified a single amino acid substitution, c.1337A>G, responsible for the specific p.Tyr446Cys missense mutation in TBL1XR1 gene.3-6,18 Since then, 2 other de novo missense mutations were found in 2 individuals with Pierpont syndrome features: c.974G > A; p.Cys325Tyr and c.1336 T > C; p.Tyr446His, both concerning the same functional and physical domains of WD40 protein. 19 The same p.Tyr446Cys sequence variant was identified in our patient. He displayed the previously reported classic Pierpont phenotype, but also some additional features, namely, significant AMC. Although several other malformations have been previously described, a subcapsular cataract has also not been mentioned before.3-7
Decreased fetal movement during intrauterine development, starting as early as 8 gestational weeks, is thought to be AMC’s mainspring.9,11,12,15,20 A wide spectrum of different underlying diseases with extrinsic (extra fetal) or intrinsic (fetal) etiologies may be involved.9,11,12 Since its first description as a congenital myodystrophy by Otto in 1841, great efforts have been made in establishing an universal definition and reaching each individual underlying etiology, with different proposed diagnostic approaches.8,9,13,21 However, due to its phenotypic heterogeneity, consensual recommendations are still not defined.
Besides clinical evaluation, with characterization of affected joints distribution and examination of craniofacial features, central nervous system and other organs investigations should be complete. Biochemical, electrophysiologic, imaging, and targeted genetic investigations are regularly performed, but frequently are not sufficient for a definitive diagnosis. 13 In those occasions, broader genetic testing is recommended. As in our case, whole exome sequencing (WES) and whole genome sequencing (WGS) have been emerging as an effective alternative for diagnosis establishment, presenting an high diagnostic yield, up to 60%. 13 In Pierpont syndrome, initial reported cases presented normal high-resolution aCGH or Single Nucleotide Polymorphism (SNP) array, which may have contributed to the lack of knowledge about its etiology during the 18-year period after its first description.
Our patient highlighted the complex clinical difficulties of both entities, the recent and limited clinical recognition of Pierpont syndrome, and the challenging diagnostic investigation of AMC, related to its multiple and heterogeneous underlying etiologies. Despite the recognition of the newborn’s distinct physical features, which led to a syndromic disorder suspicion, a targeted NGS panel was not sufficient for accurate diagnosis. In the reported case, CES proved to be an essential clarifying tool. It reinforces the carrying out of a broader genetic testing in an earlier AMC’s investigation stage, specially in atypical cases such as the one presented. Besides diagnostic purposes, it may also influence patients’ prognosis and management and allow family members’ counseling by establishing the mode of inheritance and risk of recurrence.22-24
Our case stands out for reporting the novel AMC presentation in a Pierpont syndrome newborn. He supports the relation between the p.Tyr446Cys sequence variant in TBL1XR1 gene with this rare syndrome, reinforcing its association with a distinctive and recognizable phenotype as well as expanding its clinical features to include AMC. Additional patients’s reports are needed for a better comprehensive phenotypic delineation of Pierpont syndrome and, consequently, better clinical recognition.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Verbal informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.
Prior Publication Disclosure
The authors declare that this work was not previously published in any form.