
Abstract
Autoantibodies to interferon γ, part of the first line of defense in the human immune response, constitutes a rare form of an acquired immunodeficiency in HIV-uninfected adults that can predispose to disseminated atypical mycobacterial infection. Particularly, this has been described in people of Southeast Asian origin. In this case report, we describe a previously healthy, Laotian man who presented with skin lesions consistent with Sweet syndrome that were later found to be precipitated by disseminated atypical mycobacterial disease. Extensive immunological workup revealed the patient to have autoantibodies to interferon γ, rendering him susceptible to this infection. Our report demonstrates a complex case with a multilayered diagnosis, while inviting perspective from multiple specialties. This enigmatic case emphasizes the importance of a broad differential with special attention to demographics while demonstrating the difficulty in treating certain atypical infections that are inherently multidrug resistant.
Case Presentation
A 63-year-old man presented with 2 months of cough, fever, intermittent night sweats, and 10 lbs of unintentional weight loss. Computed tomography scan showed pulmonary opacities and indeterminate mediastinal and hilar lymphadenopathy. Levofloxacin was administered without improvement, so a bronchoscopy with endobronchial ultrasound was performed. Needle aspiration of a lymph node yielded necrotic tissue without evidence of malignancy. Bacterial culture of bronchoalveolar lavage had no growth; stain for acid fast bacilli (AFB) was negative. Despite continued antibiotic administration, the fevers persisted. Skin lesions appeared, as well as pain and swelling of his wrists, elbows, knees, and ankles. A pustule developed at the site of a tuberculin purified protein derivative skin test 72 hours after placement. Two skin biopsies were nondiagnostic. The patient was transferred to our hospital for further evaluation.
The patient was born and raised in Laos where he was an irrigation engineer. He and his family immigrated to the United States decades ago and settled in Northwest Arkansas where he worked as a school custodian. His hobbies included gardening. He was not sexually active, and he never used illicit drugs. His last international travel was 6 years ago when he visited Laos and Japan.
On arrival to our hospital, the patient complained of pain related to swelling of his extremities and skin lesions. He did not have cough or other respiratory symptoms. Temperature was 102.8 °F, blood pressure was 163/85 mm Hg, heart rate was 118 beats per minute, and respiratory rate was 18 breaths per minute. There were no ocular abnormalities, oral lesions, or lymphadenopathy. Skin examination was remarkable for dusky, hemorrhagic pustules, bullae that were most prominent on bilateral proximal palms, and erythematous papules and pustules on bilateral arms, knees, ankles, and dorsal feet, with increased density proximal to joints (Figures 1 and 2). There was tight edema of all distal extremities. Previous skin biopsy sites had purulent drainage. This finding and the reaction to intradermal injection of tuberculin PPD suggested pathergy, an exaggerated inflammatory response to local skin injury.

Lesions of the hand.

Lesions of the leg.
Laboratory results showed white blood cell count of 16.2 K/mm3 with 84% polymorphonuclear leukocytes, C-reactive protein of 15.9 mg/dL, and erythrocyte sedimentation rate of >130 mm/h. Numerous blood cultures had no growth. Culture of purulent material from the site of previous skin biopsy was negative. Aspiration of the left ankle yielded 1 cc of blood-tinged fluid, which was not of sufficient quantity to send for analysis. Fourth-generation HIV testing was nonreactive. Multiple expectorated sputum specimens stained negatively for AFB and tested negatively for Mycobacterium tuberculosis DNA by polymerase chain reaction.
Diagnosis and Management
The differential diagnosis included vasculitis, reactive arthritis, hematologic malignancy, and various infections including tuberculosis, disseminated nontuberculous mycobacterial infection, the 2 mycoses that are endemic in Arkansas—blastomycosis and histoplasmosis—and one that is endemic in Southeast Asia—talaromycosis. Additionally, Sweet syndrome, Behçet disease, and pyoderma gangrenosum were considered because of their association with pathergy.
The dermatology service performed a skin biopsy. Histologic sections of the biopsy showed marked papillary edema and a dense neutrophilic infiltrate with admixed histiocytes within the papillary and superficial reticular dermis without evidence of vasculitis or leukocytoclasis (Figure 3). Special stains did not reveal fungal organisms or AFB. The patient’s clinical presentation and histopathologic findings were consistent with Sweet syndrome, also known as acute febrile neutrophilic dermatosis (Table 1), 1 which is usually precipitated or preceded by a parainflammatory process such as infection, malignancy, or autoimmune disease. In our case, the patient’s fever, cutaneous lesions, and edema all responded to treatment with prednisone and dapsone, and he was discharged with plans for close follow-up.
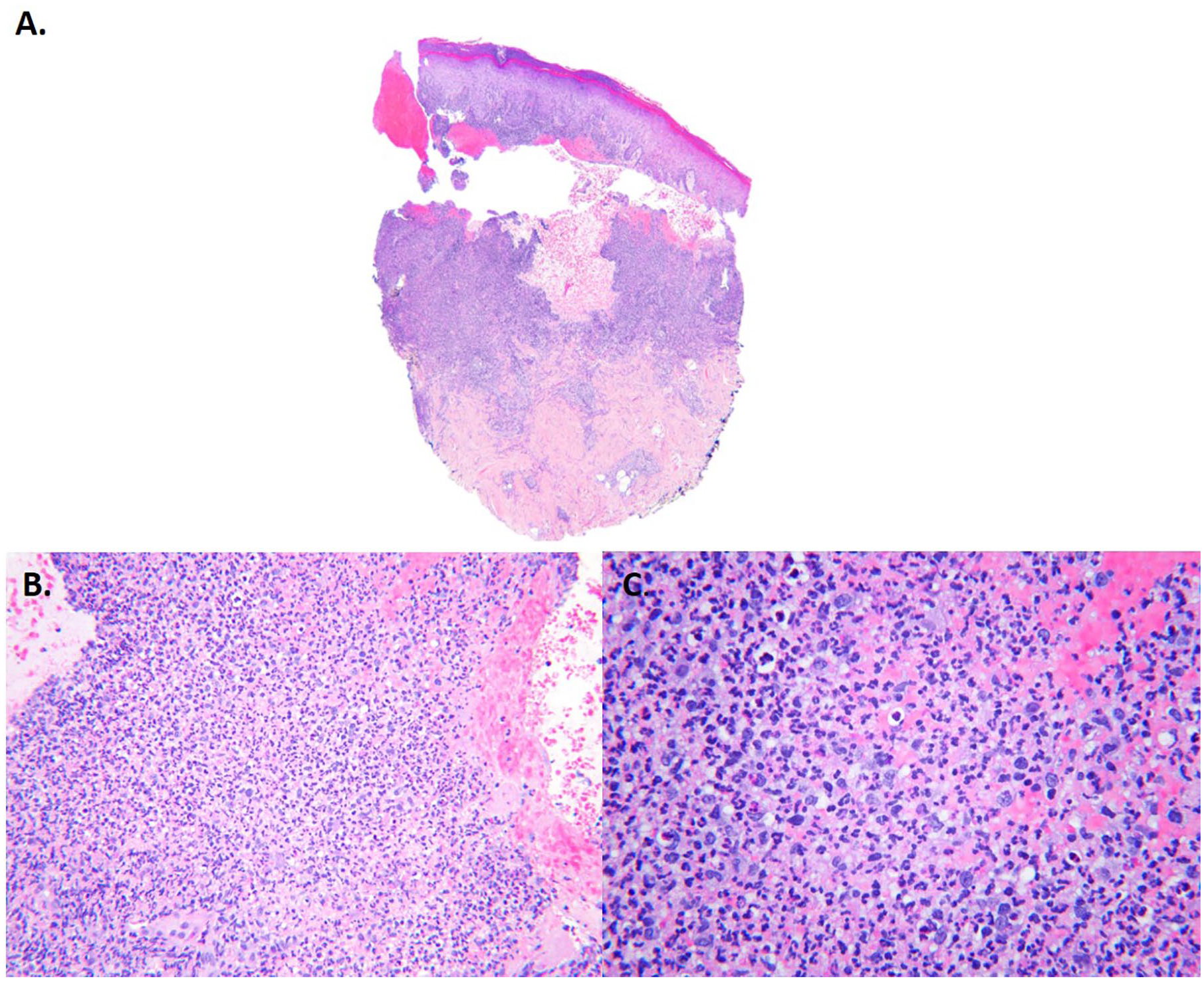
Skin biopsy (hematoxylin & eosin; A, 20×; B, 200×; C, 400×). See text for description.
Modified Diagnostic Criteria for Sweet Syndrome 1 .

Over the ensuing weeks, all signs and symptoms worsened whenever the dose of prednisone was decreased below 20 mg/day. Bacterial, mycobacterial, and fungal cultures from the skin biopsy all returned negative. Polymerase chain reaction testing of the tissue did not detect bacterial, mycobacterial, or fungal DNA. At the initial diagnosis of Sweet syndrome, no underlying trigger was identified; but 2 months later, a microbiology report from the patient’s primary care physician revealed that a culture, from a previously obtained bronchoalveolar lavage, had grown Mycobacterium abscessus/chelonae (not otherwise differentiated). Communication of the result was delayed because the community hospital sent the isolate to a reference laboratory for identification and susceptibility testing, and the significance of the result was not appreciated at that time.
About 4 months after discharge, the patient developed cervical and supraclavicular lymphadenopathy. The nodes were approximately 1 to 2 cm, mildly tender, rubbery, and not fixed. A cervical lymph node was excised; histopathology showed marked, reactive paracortical hyperplasia with focal microabscesses without granulomas, vasculitis, or malignancy. Special stains for microorganisms, including AFB, were negative, but many colonies of Mycobacterium abscessus subsp abscessus grew in culture.
Mycobacterium abscessus subsp abscessus is a rapidly growing mycobacterium that has in vitro resistance to many antibiotics. The usual therapeutic approach for pulmonary disease, which is the most common form of infection with this bacterium, includes a combination of intravenous and oral antibiotics given for more than 6 months. 2 Amikacin, which is the most active agent, is the cornerstone of therapy for M abscessus subsp abscessus infections, despite potential nephrotoxicity and ototoxicity. 2 In our case, the isolate was resistant to macrolides due to the presence of a functional erm41 gene.2,3 The patient was treated for disseminated infection with 4 months of intravenous antibiotics consisting of amikacin, imipenem, and tigecycline, plus oral clofazimine, an anti-leprosy drug procured through the Food and Drug Administration via a single patient Investigational New Drug process. 4 The patient tolerated 4 months of this potentially toxic and unwieldy regimen remarkably well. All signs and symptoms resolved. Once the infection was controlled, prednisone was tapered off successfully.
Discussion
Why did this patient, who was not known to be immunocompromised, develop disseminated mycobacterial infection? A previously unrecognized clinical entity of cervical lymphadenitis due to rapidly growing mycobacteria and reactive skin disease, that is Sweet syndrome, among adults in northeastern Thailand was reported in 2000. 5 Additional investigations supported the existence of a non-HIV-related, adult-onset immunodeficiency syndrome in Southeast Asians.6,7 People with this disorder produce neutralizing anti-interferon γ autoantibodies, which increases the risk of mycobacterial and other opportunistic infections.7-10 Serum from our patient tested positive for the presence of these autoantibodies.
When evaluated 12 months after completion of antibiotics, he was doing well without recurrence of constitutional symptoms, cutaneous disease, or cervical lymphadenopathy. After extensive evaluation of this challenging case, we concluded that the patient’s initial diagnosis of Sweet syndrome was likely precipitated by disseminated M. abscessus subsp. abscessus infection to which he was predisposed by immunodeficiency due to anti-interferon γ autoantibodies. The complexity of this case demonstrates the importance of evaluating uncommon causes of acquired immunodeficiency in seemingly healthy patients with attention to the demographics and associated predilection for disease.
Footnotes
Acknowledgements
We thank Sara Shalin, MD, and Jerad Gardner, MD, for their assistance with the histologic images and description of pathologic findings.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Verbal informed consent was obtained from the patient(s) for their anonymized information to be published in this article.