
Abstract
B-cell prolymphocytic leukemia (B-PLL) is a rare leukemia characterized by rapidly increasing leukocytosis with splenomegaly and lymphadenopathy. Treatment strategies are largely based on studies of chronic lymphocytic leukemia (CLL). Antibodies against the cell surface protein CD20 are considered to be first-line therapy. A 76-year-old male with known CLL presented 2 weeks after starting chemoimmunotherapy for newly refractory CLL after failing ibrutinib therapy. White blood cell count was elevated at 226.7 × 103/µL. Fluorescent in situ hybridization analysis of a bone marrow specimen showed new development of complex cytogenetics. Flow cytometry revealed B cells appearing slightly dimmer on CD45 and brighter on CD20 compared with typical B-CLL suggestive of less mature lymphocyte forms. The patient was diagnosed with B-PLL and started on obinutuzumab and venetoclax with rapid normalization of white blood cells. This case recapitulates the challenges in diagnosing and treating B-PLL. Ibrutinib resistance is a growing area of study with several proposed mechanisms of acquired resistance. The pathogenesis of B-PLL is not completely understood, although mutations in MYC are presumed to play a role.
Introduction
B-cell prolymphocytic leukemia (B-PLL) is a rare chronic leukemia accounting for <2% of all lymphoid leukemias. 1 It is characterized by rapidly increasing leukocytosis with splenomegaly, varying lymphadenopathy, and often accompanied by cytopenias. B-PLL is a diagnostically distinct entity when compared with chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL). However, in cases of atypical CLL and, on the rare occasion, of transformation from CLL to B-PLL, the immunophenotype overlaps significantly leading to diagnostic challenges. The key distinguishing characteristics of B-PLL is the presence of >55% prolymphocytes in blood or bone marrow (BM).2,3 B-PLL has characteristic bright CD20 expression compared with diminished expression observed in CLL. 4 Aberrations in MYC have been reported to be a common event in PLL. 5
Here we present a rare case of B-PLL transformation in a patient with progressive CLL despite appropriate therapy with ibrutinib and highlight the treatment challenges that may be encountered.
Case Presentation
A 76-year-old male was referred to the hospital by his primary oncologist due to profound leukocytosis, which developed 2 weeks after starting chemoimmunotherapy for newly refractory CLL. CLL was diagnosed in the patient 2 years and 8 months prior to this presentation on the basis of concordant immunophenotype of monoclonal B-cell population in peripheral blood and BM co-expressing CD5, CD19, CD20, CD23 (partial), FMC7, kappa light chain, negative for CD10, CD25, and CD38. Routine fluorescent in situ hybridization (FISH) studies were negative for t(11,14), trisomy 12, and deletions 11q, 13q, and 17p. The patient was determined to be intermediate risk given Rai Stage II disease and per the International Prognostic Index classification by virtue of his age and presence of splenomegaly. He was initially managed with active surveillance for 2 years until he developed progressive anemia, thrombocytopenia, and worsening leukocytosis 9 months prior to this presentation, which required initiation of treatment (see Figure 1). He was started on ibrutinib and rituximab. The patient developed fever, chills, and weakness during rituximab infusion, which was treated with antipyretics and antihistamines, and he was discharged home from the outpatient infusion center. However, over the course of the next 5 days, the patient developed decreased urinary output, fever, lower abdominal pain, and generalized weakness. He was admitted to the hospital with acute renal failure and diagnosed with tumor lysis syndrome (TLS). He was treated with intravenous fluids, allopurinol, and rasburicase and required 2 treatments of hemodialysis. Fortunately, he made full recovery of his renal function. TLS was presumed to be a consequence of the rituximab, and as such, it was discontinued and the patient was maintained on monotherapy with ibrutinib. Nine months into treatment, after initial resolution of leukocytosis, the patient again developed a progressive increase in white blood cell count with marked absolute lymphocytosis. In light of this, the ibrutinib was discontinued in favor of bendamustine and ofatumumab. The patient developed a similar infusion reaction to ofatumumab as to rituximab and was treated similarly. Within 2 weeks of treatment, there was another rapid increase in the patient’s white blood cell count resulting in the current hospitalization.

White blood cell count trend as a response to therapy.
On initial presentation, the patient was afebrile, hemodynamically stable, and in no acute distress. The cardiopulmonary examination was unremarkable. There was hepatosplenomegaly appreciated on abdominal examination without abdominal or costovertebral angle tenderness. There was no enlarged cervical, axillary, or inguinal lymph nodes detected. The rest of the physical examination was unremarkable. Laboratory values are as shown in Tables 1 and 2.
Metabolic Panel.

Abbreviations: BUN, blood urea nitrogen; LDH, lactate dehydrogenase; ALP, alkaline phosphatase; AST, aspartate transaminase; ALT, alanine transaminase.
Complete Blood Count.

Abbreviations: WBC, white blood cell; Hb, hemoglobin; Segs, segmented neutrophils; Lymphs, lymphocytes; Monos, monocytes; Eos, eosinophils.
Interval computed tomography scan of the thorax-abdomen-pelvis showed new perihilar and mediastinal adenopathy, hepatomegaly, and markedly enlarged spleen compared with prior imaging (Figure 2). A peripheral blood smear demonstrated many mature-appearing lymphocytes (Figure 3).

Noncontrast computed tomography abdomen showing marked splenomegaly with inferior displacement of the left kidney. There is extensive pericaval, periaortic, and mesenteric adenopathy. Incidental calcification of abdominal aorta consistent with atherosclerotic disease.

Peripheral blood smear showing an increased number of lymphocytes. Both small, mature lymphocytes and larger cells with less condensed nuclear material are seen. Numerous acanthocytes are present. This could be a consequence of congestive splenomegaly, marrow infiltration by tumor (myelophthisis), or both.
The patient was started on intravenous fluid hydration. In light of the patient’s white blood cell count increasing at an alarming rate, hydroxyurea was started and titrated to 1500 mg twice daily as a temporizing measure to slow the disease process and prevent complications of leukostasis. Flow cytometry of a BM aspirate revealed 87% lymphocytes of which 94% represented B-lymphocytes (immunophenotype positive for CD19, CD20, and CD5, and negative for CD10) with a kappa–lambda ratio >100. The B cells appeared slightly dimmer on CD45 and brighter on CD20 compared with typical B-CLL suggestive of less mature lymphocyte forms and possible transformation to PLL, respectively (Figure 4). The neoplasm involved approximately 50% of the core cellularity (Figure 5). FISH analysis, having demonstrated normal cytogenetics at diagnosis, now revealed deletion of both the ATM gene locus and the CCND1 gene locus suggestive of monosomy 11q. This, in addition to rearrangements of 7q and 11q, loss of chromosome 2, translocation between 1 and 7, rearrangements of 3q and 8q are all consistent with complex karyotype—generally an adverse prognostic marker in CLL, although less well described in PLL. Ki-67 immuno-stain showed an increased proliferation index. On the basis of these findings, the patient was diagnosed with PLL and started on obinutuzumab and high-dose methylprednisolone while hospitalized. Due to previous reactions to antibodies, the initial dose was split between 2 separate days and given at a slow infusion rate. Additionally, the patient was pretreated with methylprednisolone, acetaminophen, and diphenhydramine 1 hour prior to infusions. The patient responded rapidly to the medication but unfortunately developed significant thrombocytopenia requiring transfusion with 3 units of platelets while hospitalized. The patient was discharged with plans to continue monoclonal antibody therapy with obinutuzumab and the addition of the oral BCL-2 inhibitor, venetoclax, in the outpatient setting. We are pleased to report near-normalization of peripheral counts and overall excellent response now 2 months after starting this regimen.

Immunohistochemical CD20 antibody stain highlighting expression in the predominant population of B lymphocytes.
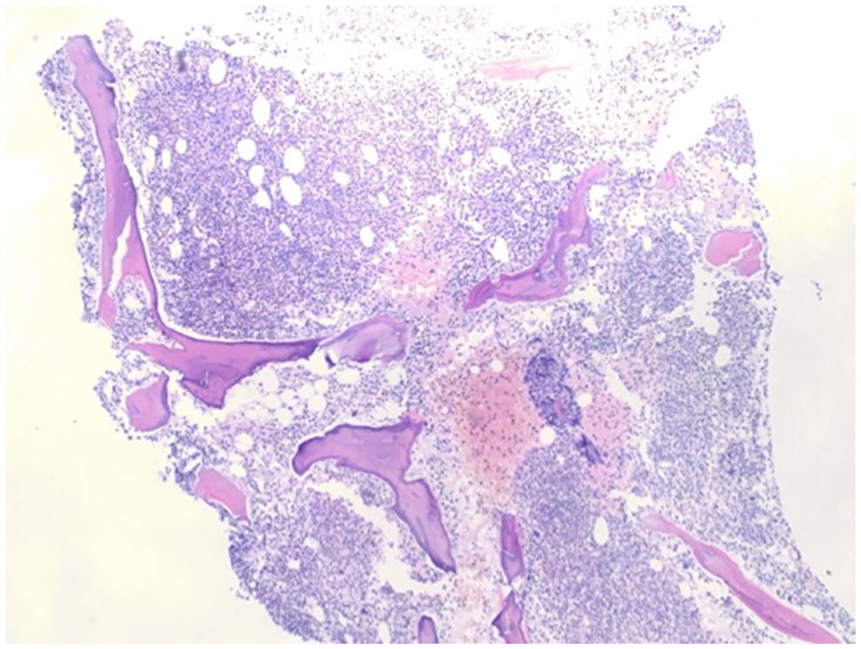
Bone marrow aspirate with marked hypercellularity involving approximately 50% of the core cellularity.
Discussion
This case highlights several challenges that can present regarding the diagnosis and treatment of B-PLL. There is a paucity of data related to the management of PLL, and consequently, treatment strategies are largely extrapolated from studies of CLL.6,7 Patient age, comorbidity profile, fitness level, as well as prognostic risk factors, including the presence of high-risk cytogenetic features (ie, deletion 17p, TP53 mutation, and complex cytogenetics), helps in selecting optimal treatment regimens. 7 Increased CD20 expression emphasizes the potential for targeted immunotherapy in its management, as with rituximab, or the glycoengineered anti-CD20 monoclonal antibody, obinutuzumab. Alkylating agents have been used with limited success and often brief responses. B cell receptor inhibitors, such as the Bruton tyrosine kinase (BTK) inhibitor, ibrutinib, are often utilized in those immunophenotypes with 17p deletions. 8
This patient unfortunately developed reactions to multiple antibodies targeted against CD20, which limits treatment options. The development of TLS in the setting of rituximab therapy is perhaps a clue that a more sinister disease process was occurring, as the true incidence of TLS from rituximab infusion is low, although has previously been described in a case of B-PLL.6,9 This may have been a reasonable time to consider disease re-evaluation with a BM biopsy, or peripheral flow cytometry or FISH analysis given the severe leukocytosis. The initial cytogenetics at time of diagnosis returned normal. However, FISH analysis on the second BM specimen was positive for deletion of the ATM gene locus on chromosome 11q indicative of an element of clonal mutation. Although aberrations in the ATM gene are one of the most frequent mutations seen in CLL, the prognostic value remains unclear. 10 Early progression of disease has been shown to having inferior overall survival compared with those with longer progression-free survival. 11 A biomarker that has been increasingly used to identify more aggressive disease processes is the proliferation index, Ki-67. This nuclear protein correlates with cell turnover and replication. It has been demonstrated to be a useful prognostic indicator in multiple solid organ and hematologic malignancies. The increased Ki-67 stain observed in this case may be a consequence of disease progression or disease transformation. 12 Although his MYC status was not reported, it is estimated that half of PLL cases have MYC mutations. The patient did have rearrangement of 8q, which could have involved the MYC locus.
Of interest, the patient developed progression of disease after 6 months of therapy with ibrutinib. Ibrutinib resistance is a growing area of study and there have been several mechanisms proposed in the setting of CLL. With regard to MCL, mutations in CCND1 have been implicated. There have been reports of deletions of the CCND1 gene locus in the absence of t(11;14). Acquired resistance to ibrutinib have also been linked to mutations in the enzyme active site in BTK or its substrate phospholipase C gamma 2 (PLCG2).13,14 It remains unknown whether these mutations are present prior to initiation of therapy or are acquired later. 15 While ibrutinib has demonstrated efficacy in treatment of CLL, MCL, and PLL, it is possible that the patient acquired resistance through one of these different mechanisms.
The pathogenesis of B-PLL is not completely understood. There has been increasing evidence in support of increased expression of MYC playing a role in the disease process similar to Burkett’s lymphoma. 16 This case was unique in that the patient had early treatment failure on 2 different biologics, as well as 2 separate infusion reactions. We speculate that the treatment failures may be a consequence of an indolent process transforming to a more aggressive disease.
Footnotes
Author Contributions
SB and MP contributed to clinical assessments, data interpretation, and developing treatment plans. All authors contributed to literature review and manuscript preparation. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
This case was evaluated by Sarasota Memorial Hospital Institutional Review Board and was deemed to not meet the definition of human subject research under the purview of the institutional review board according to federal regulations.
Informed Consent
Written informed consent was obtained from the patient for publication of this case.