
Abstract
Immunoglobulin G4–related disease (IgG4-RD) is a chronic fibrosing inflammatory systemic disorder that has been recognized relatively recently in the medical literature. Little is known about the exact disease pathogenesis and epidemiology. IgG4-RD may be asymptomatic or may have minimal symptoms or involve multiple organs with overt symptoms. The different phenotypes of IgG4-RD can lead to delayed or incorrect diagnosis. We report the case of a 66-year-old male with coal worker’s pneumoconiosis who presented with progressive kidney disease and was diagnosed with tubulointerstitial nephritis due to IgG4-RD. The patient was noted to have progressive kidney disease, skin involvement, worsening interstitial lung disease, complete vision loss in the left eye, and retroperitoneal fibrosis. Serologic workup revealed elevated inflammatory markers, IgG4 and IgG1 levels, and hypocomplementemia. A tissue biopsy helped us establish a definitive diagnosis of IgG4-RD and initiate treatment with glucocorticoids to prevent further progression of kidney disease and other end-organ damage.
Keywords
Immunoglobulin G4–related disease (IgG4-RD) is a chronic multisystem inflammatory disorder. The exact pathogenesis of the disease is not well understood and is a relatively recent diagnostic entity.1,2 Some of the characteristic features include tissue fibrosis of involved organ systems, infiltration of lymphocytes, and IgG4-secreting plasma cells in the affected tissues. Serum immunoglobulin G4 levels may be elevated during an acute flare of the disease, although the levels may be within normal limits.1,3 Another characteristic feature of the disease is the development of mass lesions in multiple organ systems, which may have the appearance of tumors. IgG4-RD can affect almost any tissue in the body, and the fibrosing inflammatory processes in the affected sites may lead to organ dysfunction.1,2,4 Frequently, the disease responds well to glucocorticoids. Untreated IgG4-RD may lead to progression of organ dysfunction with the potential for irreversible damage. As such, early diagnosis for timely treatment is imperative in all symptomatic patients with IgG4-RD.3-5
Case Presentation
A 66-year-old male with a past medical history of well-controlled hypertension, hypothyroidism, and coal worker’s pneumoconiosis was noted to have rising serum creatinine levels on outpatient evaluation and was referred for hospitalization for further evaluation of his progressive kidney disease.
He reported generalized weakness, fatigue, nausea, occasional vomiting, anorexia, and onset of a petechial skin rash that involved both lower extremities including the anterior tibial regions, lateral aspects of the upper thighs, and the inguinal areas. He had intermittent bilateral ankle edema and bilateral pedal neuropathy for about 3 weeks prior to the presentation. He reported having mild vague chronic lower abdominal discomfort for several months prior to the presentation. He had been taking an over-the-counter analgesic powder containing aspirin, acetaminophen, and caffeine daily until 5 months prior to presentation when he was hospitalized for pneumonia and was advised to discontinue taking the analgesic powder. After he discontinued taking the analgesic powder, he started taking 2 tablets of over-the-counter ibuprofen daily for his chronic headache and low back pain.
He experienced unilateral visual disturbances in his left eye 3 months prior to admission, and on ophthalmological evaluation, he was diagnosed with posterior scleritis and was prescribed oral corticosteroids for 1 month. Despite completing the corticosteroid course, his visual disturbances continued to progress with complete loss of vision in his left eye. He preferred no further workup or treatment for his left visual loss.
Review of systems was further remarkable for chronic cough with occasional production of small amounts of brown sputum, chronic exertional dyspnea, occasional episodes of epistaxis, and unintentional weight loss of about 35 to 40 pounds in the preceding several months. For these pulmonary symptoms, he received several antibiotic courses over a 6-month period.
His family history was notable for renal disease of uncertain etiology in his sister. He used smokeless tobacco in the form of tobacco dipping, but no alcohol or illicit drug use was reported. He had occupational exposure to fine particulate matter as a coal mine worker for several years. He had no history of any episodes of pancreatitis, hepatitis, or elevated transaminases. Vital signs were within normal limits. Physical examination revealed petechial rash over both shins, upper lateral aspect of both thighs and in the inguinal areas, and trace bilateral ankle edema. He was hemodynamically stable with no uremic symptoms with a good urine output, and a functional capacity more than 4 metabolic equivalents.
A trend in his serum creatinine level months prior to current admission is described in Table 1. Workup 6 months prior to current admission is shown in Table 2.
Trend in Serum Creatinine Level Months Prior to the Current Admission.

Laboratory Evaluation Findings 6 Months Prior to Current Admission.

At the time of current admission, his serum creatinine was at 9.0 mg/dL with a non-anion gap metabolic acidosis and hyperphosphatemia due to kidney disease. Plasma potassium levels were noted to be in the normal range. He had a baseline hemoglobin in the range of 11 to 12 g/dL, and his white blood cell count was chronically elevated for the preceding 6 months in the range of 10 500 to 22 900/µL. Elevated lymphocytes, granulocytes, and monocytes were found on the differential. Blood cultures were negative. Electrocardiogram did not show any significant abnormalities. Urinalysis revealed no red blood cells, white blood cells, casts, or proteinuria. Urine sodium level was 72 mEq/L, and urine creatinine level was 50 mg/dL. Hepatitis C antibody was reactive, while viral load was undetectable; hepatitis B surface antigen was negative. Further workup revealed low complement levels with low C3 and C4, positive rheumatoid factor, negative antinuclear antibody, negative antineutrophil cytoplasmic antibody, negative cryoglobulin, elevated free kappa and lambda light chains, elevated erythrocyte sedimentation rate, and C-reactive protein (Table 3). IgG subclass evaluation is described in Table 4.
Serologic Workup Findings of Our Patient During the Current Admission.
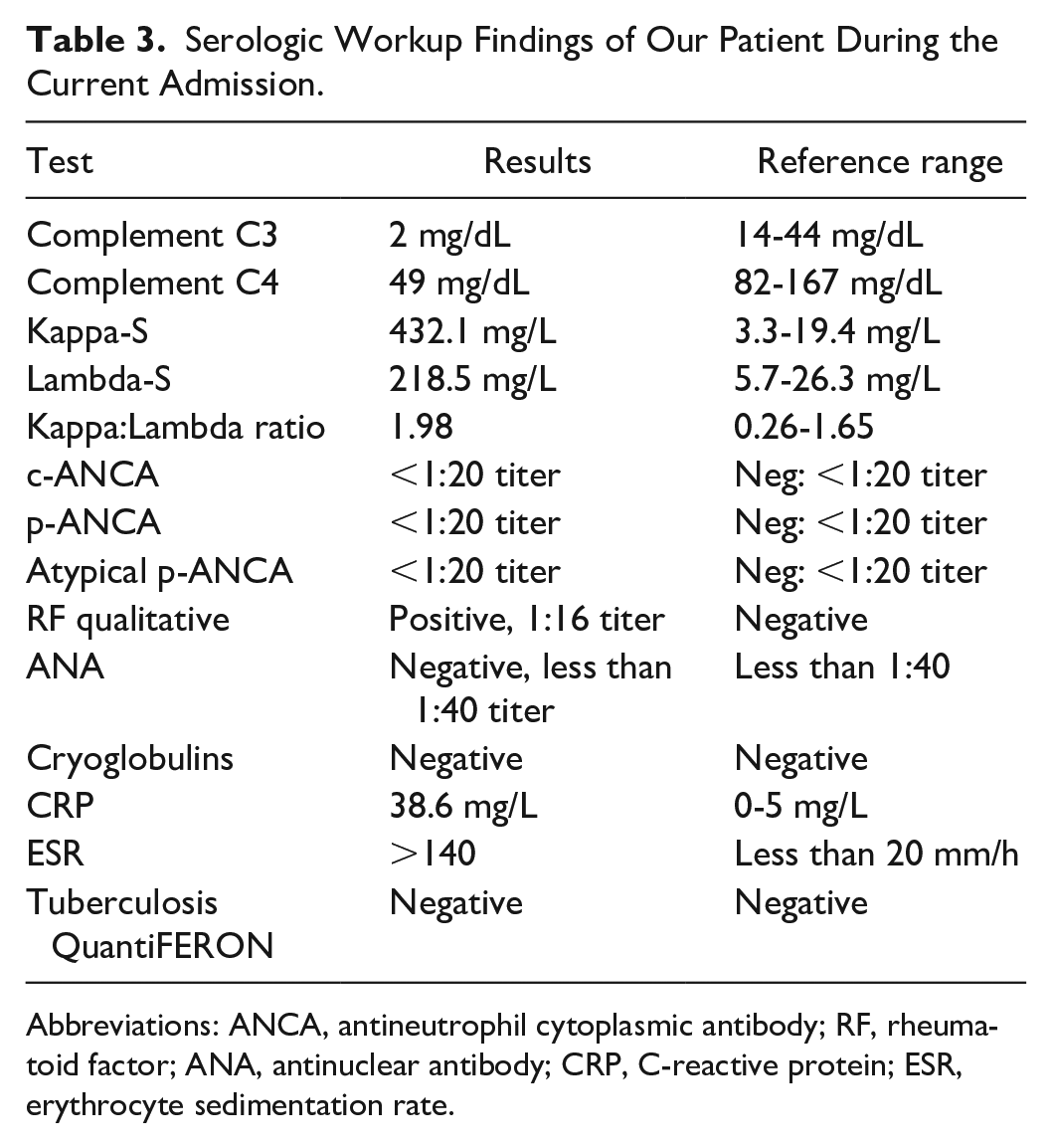
Abbreviations: ANCA, antineutrophil cytoplasmic antibody; RF, rheumatoid factor; ANA, antinuclear antibody; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate.
Laboratory Evaluation of Our Patient’s IgG Subclass Levels During the Current Admission.

Abbreviation: IgG, immunoglobulin G.
Chest X-ray demonstrated progression of interstitial lung changes, which prompted noncontrast computed tomography (CT) scan of the lungs, which revealed multiple significantly enlarged mediastinal and right hilar lymph nodes, worsened from the previous examination with changes of chronic obstructive pulmonary disease and reticular interstitial changes. Significant worsening of infiltrates predominantly in both upper lobes and right lower lobe with a small right effusion were seen (Figure 1).

Mediastinal and hilar lymphadenopathy, reticular interstitial changes.
Renal ultrasound was unremarkable. CT scan of abdomen and pelvis without intravenous contrast revealed increasing infiltrates within both lung bases and pleural thickening when compared with radiographic imaging 6 months previously, mild retroperitoneal fibrosis with prominent soft tissue in the aortocaval region below the renal vessels, nonobstructing left renal calculi, and hepatosplenomegaly without portal venous enlargement (Figures 2 and 3).

Retroperitoneal fibrosis with prominent soft tissue in the aortocaval region below the renal vessels.

Retroperitoneal fibrosis.
Nephropathological evaluation of the renal biopsy was significant for a finding of IgG4-related tubulointerstitial nephritis consistent with IgG4-RD. There was no evidence of an immune complex–mediated or active glomerulonephritis. Light microscopy of the renal biopsy showed diffuse plasma rich inflammatory cell infiltrate throughout fibrotic interstitium with a focal storiform pattern, scattered lymphocytes, and few eosinophils within the inflammatory infiltrate. Immunohistochemical staining for IgG4 was strongly positive for staining within the interstitial plasma cell infiltrate. Immunofluorescence microscopy showed granular IgG staining along tubular basement membranes.
He was administered a single bolus of 500 mg intravenous methylprednisolone and was discharged on prednisone 40 mg by mouth daily with dose tapering to be determined on future outpatient visits.
At his outpatient nephrology follow-up visit a month after discharge, his serum creatinine levels showed a steady decline to 3.0 mg/dL. There was also a decline in inflammatory markers with C-reactive protein at 6.5 mg/L and erythrocyte sedimentation rate at 43 mm/h while on prednisone 40 mg by mouth daily. He will be followed closely in the outpatient setting for further prednisone dose tapering and renal function monitoring.
Discussion
Patients with IgG4-RD may be asymptomatic or may have only mild symptoms. Frequently, patients are found to have mass lesions from the IgG-RD which can easily be mistaken for other conditions; thus, patients may be misdiagnosed for months to years.4-7 It is quite possible that mass lesions from IgG4-RD can be misdiagnosed as malignancy. 4 Multiple organ systems may be involved, and patients may develop vision loss, hearing deficits, pleural effusion, constrictive pericarditis, and chronic kidney disease. Constitutional symptoms such as weight loss and fatigue have been described and were noted in our patient. Kidney involvement in IgG4-RD may manifest in several different ways, including ureteral obstruction from retroperitoneal fibrosis to the tubulointerstitial disease with plasma cell infiltration seen in this patient.6,8-11 Patients with retroperitoneal fibrosis may complain of vague pain.11,12 The patient in the case did complain of vague lower abdominal pain for several months prior to current presentation and reported gradual worsening of his lower abdominal pain that led to a diagnosis of retroperitoneal fibrosis on CT imaging, but there were no signs of hydronephrosis or obstructive uropathy. Another way that the kidneys can become injured in IgG4-RD is from tubulointerstitial infiltration with IgG4-secreting plasma cells.
A tissue diagnosis is required for the definitive diagnosis of IgG4-RD. A tissue sample from an affected site classically shows histopathological features of tissue infiltration by lymphocytes, IgG-positive plasma cells, a storiform pattern of fibrosis, or obliterative phlebitis. Eosinophilic tissue infiltration or nonobliterative phlebitis may be observed on histopathologic examination as well.5,6,13-16
Given the protean manifestations of IgG4-RD, it has recently been postulated that a variety of other clinical conditions may represent IgG4-RD. 13 These disorders include type 1 autoimmune pancreatitis, Riedel’s thyroiditis, Mikulicz’s disease, pseudotumor at different sites, Küttner’s tumor, inflammatory aortic aneurysm, fibrosing mediastinitis, retroperitoneal fibrosis, idiopathic interstitial nephritis, or aortitis.13-15 Elevated serum IgG4 levels as seen in this patient are neither very sensitive nor specific for the diagnosis.5,8,16
The pathogenesis of hypocomplementemia observed in IgG4-RD is unclear.12,14,16 There is some indication that hypocomplementemia is a feature of IgG4-RD and that the hypocomplementemia is driven by other IgG subclasses. For instance, our patient was noted to have elevated IgG1 and IgG4 levels with low levels of C3 and C4. It might thus suggest that elevated IgG subclasses may play a role in activating the complement system. There have been reports suggesting that patients in general with hypocomplementemia may have elevated IgG subclasses when compared with patients who have normal complement levels.1,14,16,17 Hypocomplementemia may have some relationship with elevations in other IgG subclasses besides IgG4 in the disease. However, in the current literature, there is no clear understanding of which complement activation systems are involved in IgG4-RD and if they are associated with the disease pathogenesis. More research is needed to explore the association between the disease pathogenesis and hypocomplementemia, if any, which may provide more clues toward management in refractory cases.1,12,14-17
The current standard of treatment is to induce and maintain remission of symptomatic active IgG4-RD. Early diagnosis and treatment are required to prevent continued inflammation, fibrosis, and irreversible organ dysfunction. A tissue diagnosis of IgG4-RD helped us with timely goal-directed treatment with glucocorticoids resulting in marked improvement in kidney function, thereby preventing progression to end-stage renal disease. Currently, initial therapy for active disease is glucocorticoids, though optimal initial dose and tapering schedule are unknown. It may lead to improvement in symptoms and in radiologic and laboratory markers.15,18,19 Disease recurrence after glucocorticoid taper is common. If patients have severe disease manifestations leading to organ dysfunction or failure or have a high risk of relapse, then a maintenance therapy becomes essential, which may be with a low dose of glucocorticoid or a steroid-sparing agent.16,18-20
Conclusion
The disease pathogenesis, epidemiology, incidence, and prevalence of IgG4-RD is not well known in the medical literature as it is a relatively recently discovered pathological entity. Early diagnosis and prompt treatment are of great importance to reduce the risk of progression and the development of permanent organ damage. Our patient was found to have hypocomplementemic tubulointerstitial nephritis leading to progressive renal failure. A correlation of the entire clinical presentation and laboratory and radiographic findings with the exclusion of other serious etiologies should be made, and if there is any suspicion of the IgG4-RD, a prompt tissue biopsy of the affected site must be made for early confirmatory diagnosis and treatment.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval to report this case was obtained from the Department of Veterans Affairs (VA FORM 0897).
Informed Consent
Verbal informed consent was obtained from the patient for their anonymized information to be published in this article.