
Abstract
Critically ill patients are known to have a variety of electrolyte abnormalities. Lactic acidosis can frequently be seen secondary to shock states and is usually treated with aggressive volume resuscitation. Interestingly, hypophosphatemia is a potential cause of resistant lactic acidosis, which may not be as commonly identified or considered. We present a case of a 42-year-old man admitted twice over a span of 6 months with an elevated lactate level that did not resolve with volume resuscitation. It was ultimately determined that his lactic acidosis was due to hypophosphatemia after ruling out other potential causes. Phosphate replacement therapy resulted in the normalization of his lactate. In the literature, multiple theories have indicated the association of hypophosphatemia with lactic acidosis though no prior cases exist supporting a direct relationship. In this case, we set forth to evaluate the complicated relationship between all of these factors and to highlight the importance of early detection and treatment of hypophosphatemia, which may be beneficial in treating lactic acidosis.
Introduction
Electrolyte disturbance is common in critical illness with various physiological pathways involved. Lactic acidosis and hypophosphatemia are among these disturbances, with the latter having an incidence of up to 34%. Importantly, they can have consequences affecting morbidity and mortality.1,2 Understanding the underlying involved mechanism is essential for proper diagnosis and management, especially with regard to lactic acidosis, as it can present secondary to different mechanisms, and hypophosphatemia is underrecognized potential cause.
Case Presentation
A 42-year-old male with a history of chronic alcoholism and alcoholic liver cirrhosis presented to the emergency department with epigastric pain. He reported no smoking or illicit drug use. On presentation, he was afebrile with a blood pressure of 130/80 mmHg, heart rate of 122 beats per minute, respiratory rate of 18 breaths per minute, and oxygen saturation of 98% on room air. His physical examination was remarkable for scleral icterus, abdominal ascites, and epigastric tenderness. Initial laboratory evaluation was remarkable for lactic acidosis of 9.8 mmol/L, hypomagnesemia of 1.4, elevated B-hydroxybutyric acid of 53.9 mg/dL, low bicarbonate level of 8 mmol/L, and venous pH of 7.27, as shown in Table 1. Chest X-ray was unremarkable, and computed tomography scan of the abdomen showed findings of chronic pancreatitis. He was admitted to the intensive care unit for management of suspected starvation ketoacidosis and lactic acidosis, which was thought to be secondary to alcoholism/underlying liver cirrhosis. He was administered aggressive volume resuscitation and electrolyte replacement, as shown in Table 2. His markedly elevated lactate on admission of 9.8 mmol/L decreased to 5.1 mmol/L, yet a few hours later it increased to 9 mmol/L. During this same time period, his phosphate level dropped from 2.5 mg/dL to 1.1 mg/dL. At that point, phosphate replacement was initiated without further intravenous fluid and with the correction of phosphate levels, the lactate normalized, as shown in Figure 1. He was discharged home in stable condition on multivitamins, magnesium oxide, thiamine, and folic acid. Five months later, he presented to the emergency department with encephalopathy secondary to hypoglycemia with a glucose level of 26 mg/dL that was thought to be largely related to poor oral intake and vomiting before presentation, which was corrected by dextrose 50%. His vital signs were stable, and his initial laboratory work was remarkable for lactic acidosis of 21.1 mmol/L, elevated β-hydroxybutyric acid of 33.6 mg/dL, low bicarbonate level of <5 mmol/L, and venous pH of 6.96, as shown in Table 2. He was started on aggressive volume resuscitation and electrolyte replacement, as shown in Table 2. Notably, his lactate again initially improved from 21.1 mmol/L on admission to 14.5 mmol/L. However, his phosphate level over that same time period dropped from 6.5 mg/dL on admission to 1.1 mg/dL. Phosphate replacement was initiated without additional intravenous fluid resuscitation. With the correction of phosphate levels, his lactate normalized, as shown in Figure 2.
Laboratory Results on Presentation on the First Admission.

Laboratory Results on Presentation on the Second Admission.

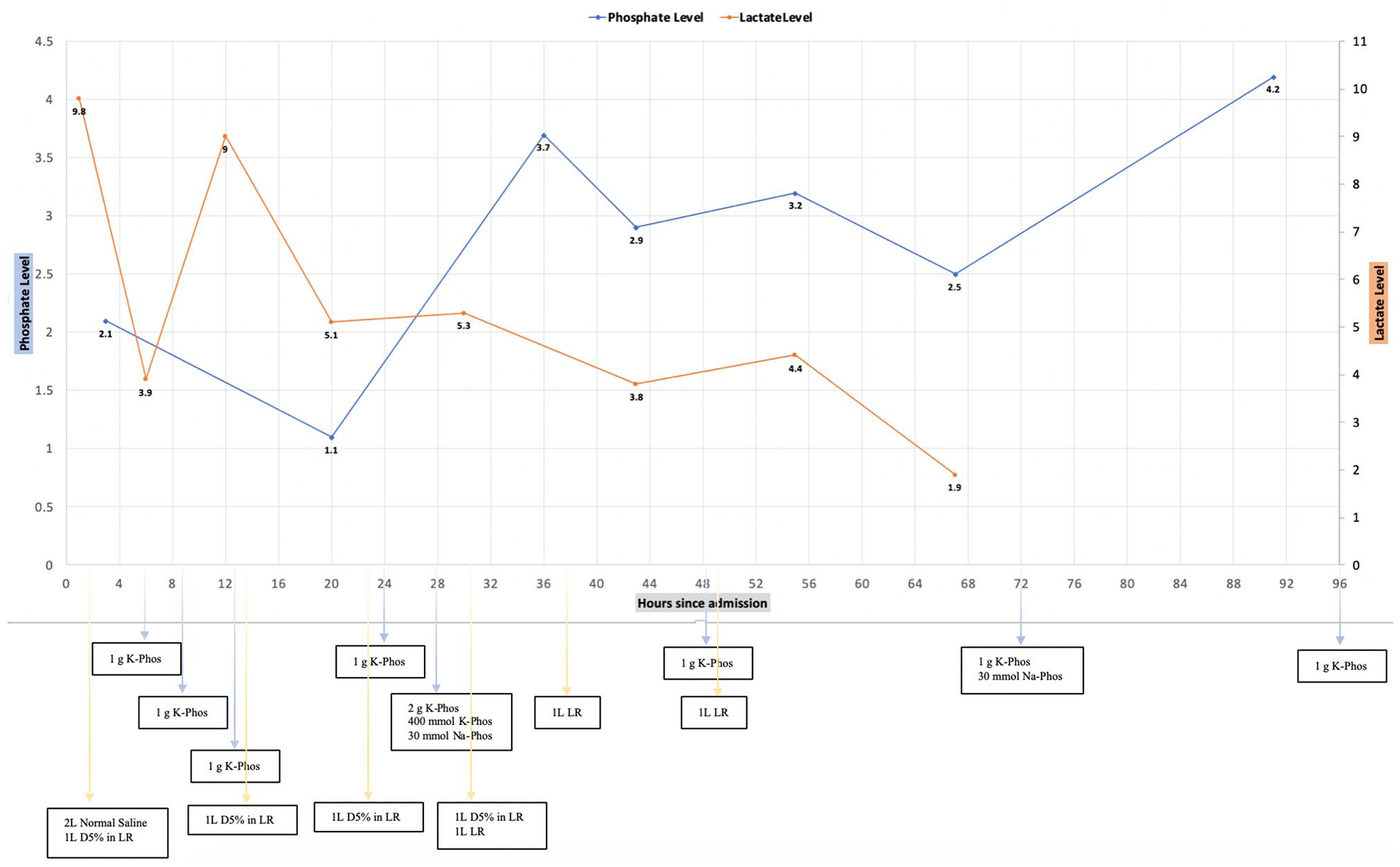
Lactate and phosphate trending throughout the first admission (K, potassium; Phos, phosphate; D5%, dextrose 5%; LR, lactate Ringer’s; Na, sodium).

Lactate and phosphate trending throughout the second admission (K, potassium; Phos, phosphate; D5%, dextrose 5%; LR, lactate Ringer’s; Na, sodium; HCO3, bicarbonate; D5W, dextrose 5% in water).
Discussion
Hypophosphatemia, defined as a serum phosphate less than 2.5 mg/dL, can be seen secondary to 3 general causes: decreased phosphate intake or intestinal absorption, increased renal excretion, or internal distribution of inorganic phosphate. 3 It is important to note that normal or even high phosphate levels can be present in total body phosphate depletion, and hypophosphatemia in the serum will not be evident until an intervention is initiated to induce the internal distribution of inorganic phosphate. Our patient’s phosphate level had been normal historically in the outpatient setting with a range between 4.0 and 4.3 mg/dL. However, during both hospitalizations, his phosphate levels on admission dropped precipitously within a few hours after volume resuscitation and administration of dextrose-containing fluid. It is thus crucial to monitor phosphate and other electrolytes carefully on several consecutive days, especially after initiation of nutrition or volume resuscitation to avoid such consequences that are referred to as refeeding syndrome, as shown in Tables 3 and 4. Also, our patient represents a good example of an important clinical entity of phosphate depletion in chronic alcoholism, which is largely related to poor phosphate intake and vitamin D intake, which in turn leads to decreased phosphate renal reabsorption and intestinal absorption. 4
Electrolytes Trend Throughout the First Admission Along With Total Electrolytes Replacement Therapy Administered in a 24-Hour Period.

Abbreviations: K, potassium; Phos, phosphate; Mag, magnesium; Ox, oxide; Na sodium.
Electrolytes Trend Throughout the Second Admission Along With Total Electrolytes Replacement Therapy Administered in a 24-Hour Period.

Abbreviations: K, potassium; Phos, phosphate; Mag, magnesium; Ox, oxide; Na, sodium.
Internal distribution of inorganic phosphate is the most common cause of hypophosphatemia in critically ill patients and is of particular importance among alcoholics, such as our patient, who are admitted to the hospital and then experience a fall in their serum phosphate within a few hours. 5 This is largely related to intracellular transition of phosphate induced by insulin release as a response to dextrose-containing fluid administration, refeeding, or respiratory alkalosis that can occur with a hyperventilatory state in alcoholics. 6
Lactic acidosis may be seen in critically ill patients under many circumstances and is generally associated with poor clinical outcomes, particularly with sepsis and low-flow state. 2 The causes can be stratified according to whether tissue hypoxia is present or absent (Figure 3). 7 Also, it important to note the benign nature of elevated lactate that might happen with infusion of large volume of Ringer’s lactate. That increase in lactate is usually minimal, however, it can be significant in cases of decreased lactate clearance.7,8 For example, decreased lactate clearance in liver dysfunction states, as the liver accounts for 70% of total body lactate clearance. 7

General scheme of causes of lactic acidosis.
Lactic acidosis should be viewed within the clinical context as the approach may differ depending on the etiology. Our patient had received a large volume of intravenous fluids despite the absence of clinical signs of shock or low-flow state. Given the persistence of his lactic acidosis despite this intervention, evaluation for other potential etiologies was sought. Although we suspected his underlying chronic alcoholism and cirrhosis were contributing, we also hypothesized that persistent lactic acidosis was related to hypophosphatemia.
Various pathophysiological mechanisms exist to explain hypophosphatemia as a potential etiology, as outlined in Figure 4. Depletion of inorganic phosphate and adenosine triphosphate (ATP) with a fall in 2,3-diphosphoglycerate lead to decreased tissue oxygen delivery due to changes in hemoglobin, oxygen dissociation impairment, and consequently lactate production.9,10 Furthermore, the phosphofructokinase enzyme in the liver, a central enzyme in the glycolysis process, will be stimulated by depletion in inorganic phosphate and ATP, promoting glycolysis and lactate formation. Additionally, alkalosis, which may develop with hypophosphatemia or secondary to hyperventilation in alcoholics, stimulate phosphofructokinase activity. Moreover, hepatic utilization of lactate into glucose will be impaired as it is an active process that requires 6 ATP molecules.10,11 In our patient, his lactate clearance improved as demonstrated by normalization of lactate on correction of his hypophosphatemia and not related to volume resuscitation. This finding supports the relationship between improving lactic acidosis with correction of hypophosphatemia.

Mechanisms of lactic acidosis in hypophosphatemia and chronic alcoholism.
Conclusion
The role of hypophosphatemia in resistant lactic acidosis has not been clearly established. We suggest future prospective studies to assess such a relation.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Verbal informed consent was obtained from the patient(s) for their anonymized information to be published in this article.