
Abstract
Introduction
In women who undergo mastectomy for the treatment of breast cancer, autologous reconstruction using the deep inferior epigastric perforator (DIEP) flap is a common surgical procedure that restores both the appearance and texture of the breast(s).1,2 Postoperatively, the abdominal donor site is routinely monitored for wound dehiscence (ie, all degrees of separation of the margins of a closed surgical incision), 3 which has a typical incidence of 3.5%-14%, but reported as high as 39%.4,5
In the setting of autologous breast reconstruction, abdominal wound dehiscence can result in prolonged hospital admission, delayed patient recovery and the need for repeat operative intervention.4,6 While the reasons for dehiscence are multifactorial; it is frequently secondary to tension forces across the incision line, and surgical site infection.3,4 Risk factors for abdominal wound dehiscence in DIEP flaps have been well documented and include: age > 65 years, body mass index (BMI) >30 kg/m2, type 2 diabetes mellitus, and smoking status. 7
Recent systematic reviews and meta-analyses of closed-incision negative pressure therapy (ciNPT) devices used for surgical and/or abdominal incisions demonstrate reduction in wound and overall abdominal complications when applied to closed surgical wounds.8–11 In the context of the DIEP flap, a retrospective cohort study by Siegwart et al 12 demonstrated a significant reduction in major abdominal donor site complications when ciNPT was applied over standard tape dressings [ciNPT (28.6%) versus Standard (50.4%), P = .001]. A similar finding was noted in a pilot randomized study performed by Muller-Sloof et al 4 who reported a statistically significant reduction in wound dehiscence in this patient population [ciNPT (8%) versus Standard (33%), P = .038]. While existing evidence supports the use of ciNPT in DIEP breast reconstruction, there exists a paucity of adequately powered, prospective clinical trials within the plastic surgery literature.
The primary objective of the present pilot trial was to assess the feasibility of conducting a larger randomized controlled trial (RCT) comparing ciNPT versus conventional dressings for DIEP reconstruction. The secondary objective of this study was to evaluate other clinical outcomes (wound dehiscence, seroma formation, surgical site infection, quality of life, scar appearance) comparing ciNPT to standard tape dressings applied to the abdominal donor site incision. The design and conduct of the definitive trial will mirror the methodology of this pilot trial including randomization, interventions, and clinical outcomes. 13
Methods
The study design was a parallel, between-group randomized control pilot study. This study was approved by the Hamilton Integrated Research Ethics Board (HiREB) (#13155) and registered on ClinicalTrials.gov (NCT04985552). The study was developed and reported according to the Consolidated Standards of Reporting Trials (CONSORT) 2010 statement: extension to randomized pilot and feasibility trials. 14
Study Population
A sample of consecutive patients who presented to outpatient plastic surgery clinics at Juravinski Hospital and St. Joseph's Healthcare Hamilton (Hamilton, Ontario, Canada) and consented to DIEP breast reconstruction were approached for study enrollment. A non-probability convenience sample was employed to minimize volunteerism and selection bias by consecutively selecting subjects who meet the study inclusion criteria. Study patients were referred from both academic and community-based general surgery practices. Patients were recruited from the practices of four fellowship-trained plastic surgeons who routinely perform DIEP breast reconstructions in their practice.
Participants were considered for inclusion if they were (i) adult female patients (≥18 years old), who (ii) consented to elective immediate or delayed breast reconstruction using the DIEP flap. Exclusion criteria included: (i) patients who were pregnant, (ii) had a documented/reported allergy to adhesive dressings, or (iii) had a BMI ≥ 40 kg/m2.
Randomization
Participants were randomly assigned using a central randomization service to receive ciNPT versus traditional adhesive dressings in a 1:1 ratio using randomly permuted blocks of 4 or 6 and developed by an independent biostatistician. Specifically, randomization occurred intra-operatively immediately following abdominal incision closure and was performed by an on-site research assistant.
Intervention (ciNPT)
The treatment group had their abdominal incisions sutured according to routine clinical practice: 3-0 VICRYL ® suture (Ethicon, USA) for deep fascia closure, 4-0 MONOCRYL® suture (Ethicon, USA) for deep dermal closure, and 5-0 MONOCRYL® suture (Ethicon, USA) for subcuticular closure. Following this, the closed-incision was dressed with the ciNPT system [V.A.C.ULTA™ Negative Pressure Wound Therapy System (KCI, USA)] applied by the operating surgeon in a sterile fashion. The ciNPT device was set at −125 mmHg of continuous negative pressure until patient discharge from hospital, or for a maximum period of 7 days. Removal of the ciNPT dressing was performed by the surgical team on the day of discharge. For ciNPT dressings removed before 7 days due to patient discharge, conventional tape dressings were applied to the abdominal incision to be left in situ until the 7-day mark.
Control (Standard Tape Dressings)
The control group underwent abdominal incision closure according to routine clinical practice (as described above). Following this, the closed abdominal incision was dressed with 1-inch Micropore™ Surgical Tape (3M, USA) with an alcohol swab applied to the tape for additional adhesion, by the operating surgeon in a sterile fashion. This dressing remained in situ for up to 7 days. If patients were discharged from hospital before 7 days they were discharged with tape dressings in situ. Following removal, no additional dressings were applied unless clinically indicated secondary to dehiscence.
Study Outcomes
The primary outcome(s) of this feasibility study were to evaluate patient eligibility, recruitment, and retention as per a priori defined criteria below:
Eligibility: At least 90% of screened patients were eligible. Recruitment: At least 85% of eligible patients were enrolled. To be considered fully enrolled patients must sign the informed consent form, complete baseline demographic questionnaires, and be randomized to a study arm. Retention: At least 85% of randomized patients completed the study, defined as completion of 6-month follow-up questionnaires (previously 12-month follow-up, protocol amended a priori to patient recruitment).
As per Thabane et al, 13 the authors evaluated these primary outcomes and chose to: (1) stop—main study not feasible; or (2) continue, but modify protocol—feasible with modifications; or (3) continue without modifications, but monitor closely—feasible with close monitoring; or (4) continue without modifications—feasible as is.
The secondary outcomes were (1) the between-group difference in the incidence of abdominal wound dehiscence; (2) seroma formation; and (3) surgical site infection necessitating antibiotic administration (diagnosed clinically or by wound swab). These outcomes were evaluated at 2 weeks and 6 weeks postoperatively. 4
Outcome assessment was performed by an independent, trained member of the research team who was blind to treatment allocation. Patients who did not attend follow-up appointments were contacted directly via telephone to review the patient's postoperative hospital record for evidence of the outcome within the time horizon.
Additionally, three independent, validated questionnaires were used to evaluate patient quality of life during this study period: (1) EQ-5D-5L, (2) BREAST-Q physical well-being abdomen, and (3) SCAR-Q. These scales are widely used patient-reported outcome measures (PROMs) designed to measure overall health status, 15 the negative physical sequelae of the abdomen following autologous tissue reconstruction, 16 as well as the appearance, symptoms, and psychosocial impact of scars, 17 respectively.
The BREAST-Q physical well-being abdomen and EQ-5D-5L were measured pre-operatively (at the time of study enrollment), as well as at 1-week, 3-months, and 6-months postoperatively. The SCAR-Q was measured at 1-week, 3-months, and 6-months postoperatively. This time horizon was selected based on routine patient follow-up, clinical experience regarding time to achieve a stable outcome and is in-keeping with similar trials that have evaluated this research question.4,18,19
Sample Size and Study Duration
As the purpose of this pilot study was to evaluate trial procedures and processes, rather than the efficacy of the interventions, conventional calculations of sample size are deemed to be larger than necessary. The pilot phase of the study utilized the sample size rule of thumb established by Julious 20 —a minimum sample size of 12 participants per treatment arm (n = 24).
Data Collection and Statistical Analysis
Participant demographic data, baseline EQ-5D-5L and BREAST-Q physical well-being abdomen were recorded by a trial research assistant at the time of study inclusion. Variables such as age, BMI, smoking status, presence of type 2 diabetes, and history of prior radiation or chemotherapy were recorded at baseline and summarized between-groups using descriptive statistics where appropriate.
Feasibility outcomes and clinical outcomes were analyzed using descriptive statistics. Patients were analyzed within the group they were allocated to follow the intention-to-treat principle. Inferential statistical analysis was not performed for clinical outcomes as this pilot study was not intended to measure efficacy. The incidence rates of the wound dehiscence clinical outcome will be used to calculate the final sample size of the study.
Results
There were 24 patients recruited and randomized between March 2022 and January 2023. Twelve patients were assigned to each study arm. See Figure 1 for a flow diagram of patient selection.
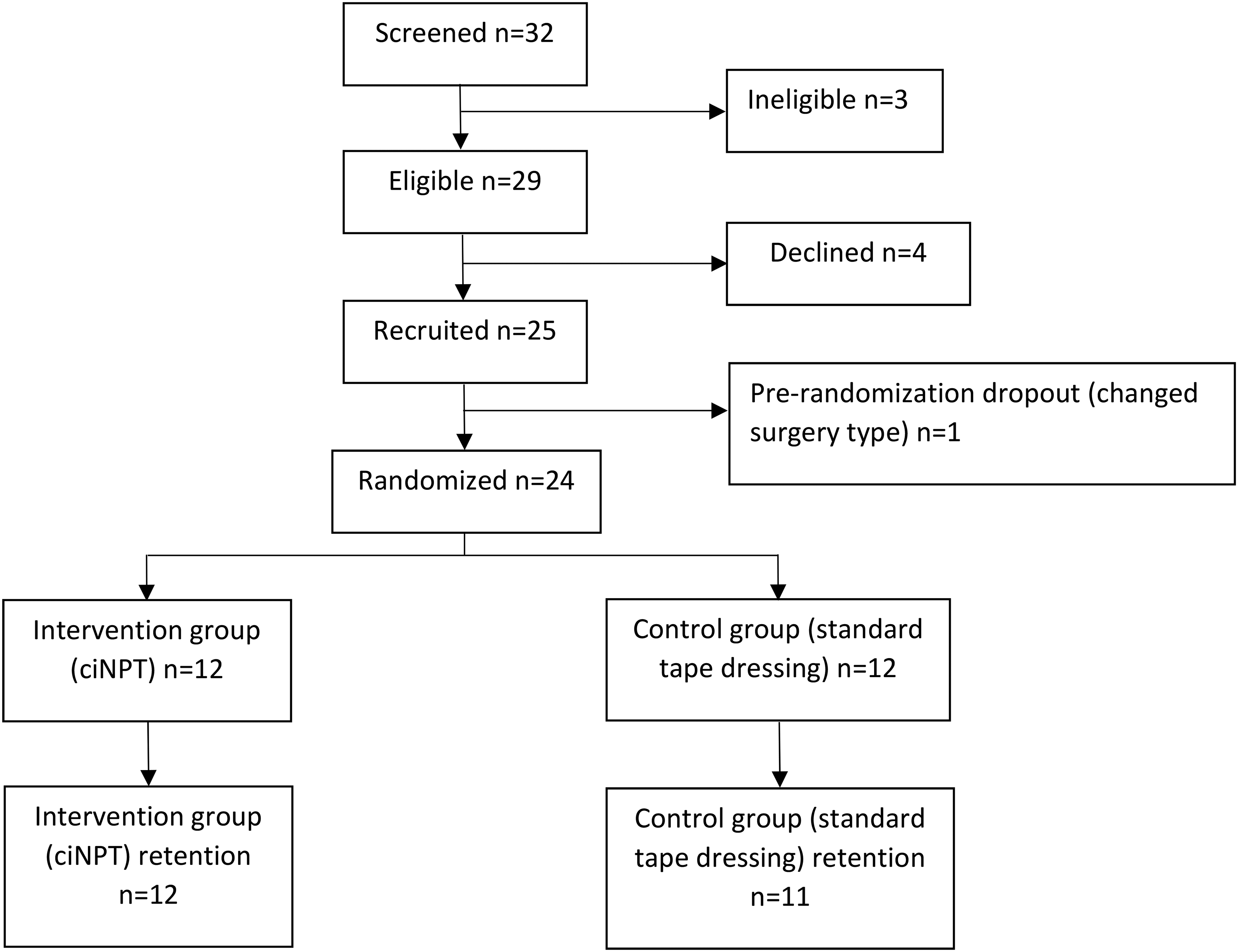
Patient flow diagram.
The mean age of included patients was 50.3 (8.8) in the intervention group and 47.2 (8.0) in the control group. Patients had a mean BMI of 31.1 (3.3) in the intervention group and 30.1 (2.8) in the control group. In the intervention group, 4/12 patients were former smokers, 1/12 patients had diabetes, 6/12 patients had radiation, and 6/12 patients had chemotherapy. In the control group, 3/12 patients were former smokers, 1/12 patients had diabetes, 6/12 patients had radiation, and 6/12 patients had chemotherapy.
There were 10 patients undergoing bilateral reconstructions (6 delayed, 3 immediate, 1 mixed) and 2 patients undergoing unilateral reconstructions (2 delayed) in the intervention group. In the control group, there were 8 patients undergoing bilateral reconstructions (4 delayed, 3 immediate, 1 mixed) and 4 patients undergoing unilateral reconstruction (3 delayed, 1 immediate).
Patient characteristics are also available in Table 1.
Patient Characteristics.

SD, standard deviation.
Feasibility Outcomes
Feasibility outcomes are summarized in Table 2. The eligibility rate was 90.6% (29 eligible of 32 screened) and recruitment rate was 86.2% (25 recruited of 29 eligible). Pre-randomization retention rate was 96.0% (24 randomized of 25 recruited; 1 dropout due to changed surgery type), and post-randomization retention rate was 95.8% (23 retained of 24 randomized). The 1 patient not retained post-randomization (in the control group) attended all follow-ups with the surgeon but did not complete any questionnaires. Based on the predetermined feasibility criteria, the full RCT was deemed to be feasible as is with the recommendation to continue without modifications.
Feasibility Outcomes.

Secondary Outcomes
Abdominal complications are summarized in Table 3 and described in detail in Table 4. Wound dehiscence rates were 8.3% (1 event) in the intervention group and 41.7% (5 events) in the control group. Seroma rates were 16.7% (2 events) in the intervention group and 8.3% (1 event) in the control group. There were no wound infections recorded. All complications were deemed to be clinically significant. Wound dehiscence was typically managed with daily dressing changes and negative pressure wound therapy was used in 4/6 patients. Two patients, both in the control group, required revision surgery for wound debridement. Seromas were all aspirated in clinic. There was 1 patient with a minor adverse reaction in the intervention group; the patient developed blisters underneath the ciNPT dressings which self-resolved.
Summary of Abdominal Wound Complications.

Details of Abdominal Wound Complications.
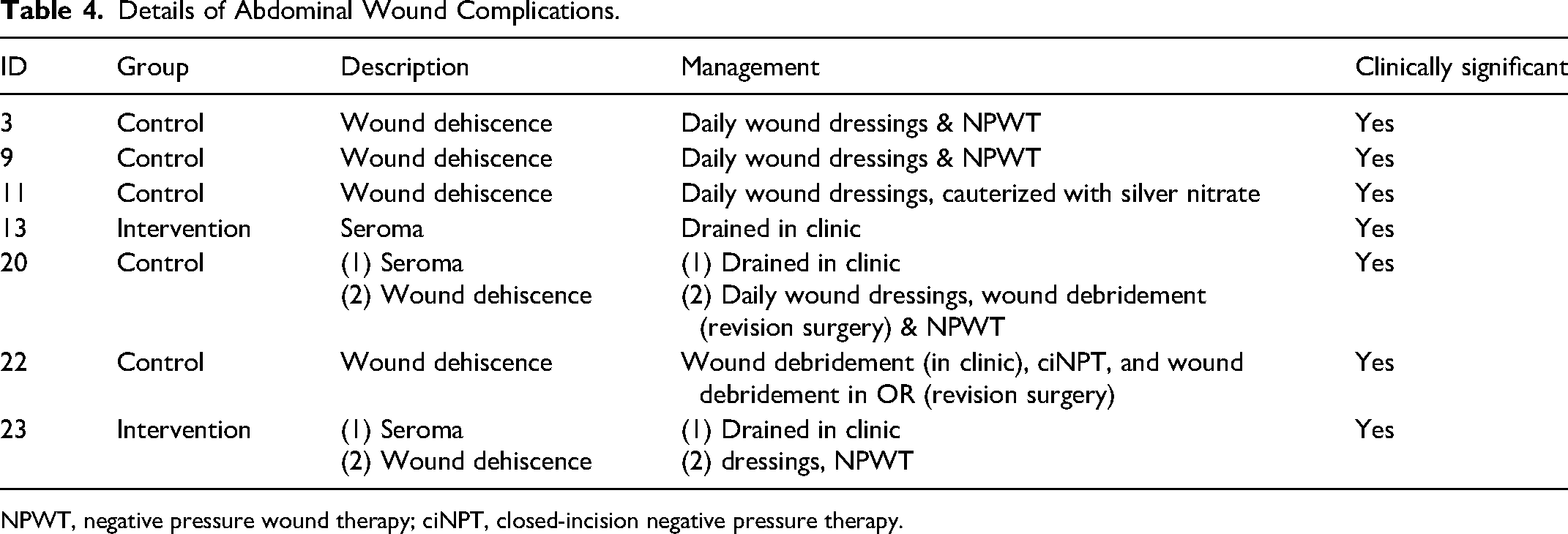
NPWT, negative pressure wound therapy; ciNPT, closed-incision negative pressure therapy.
Scores from the BREAST-Q: Physical well-being—Abdomen, EQ-5D-5L, and SCAR-Q are summarized in Table 5. One patient in the control group did not complete any questionnaires despite multiple follow-up communications. PROM completion rate was highest at baseline and 6 months (95.8%, 23/24) and lowest at 3 months (75.0%, 18/24).
Summary of Patient-Reported Outcome Measures.
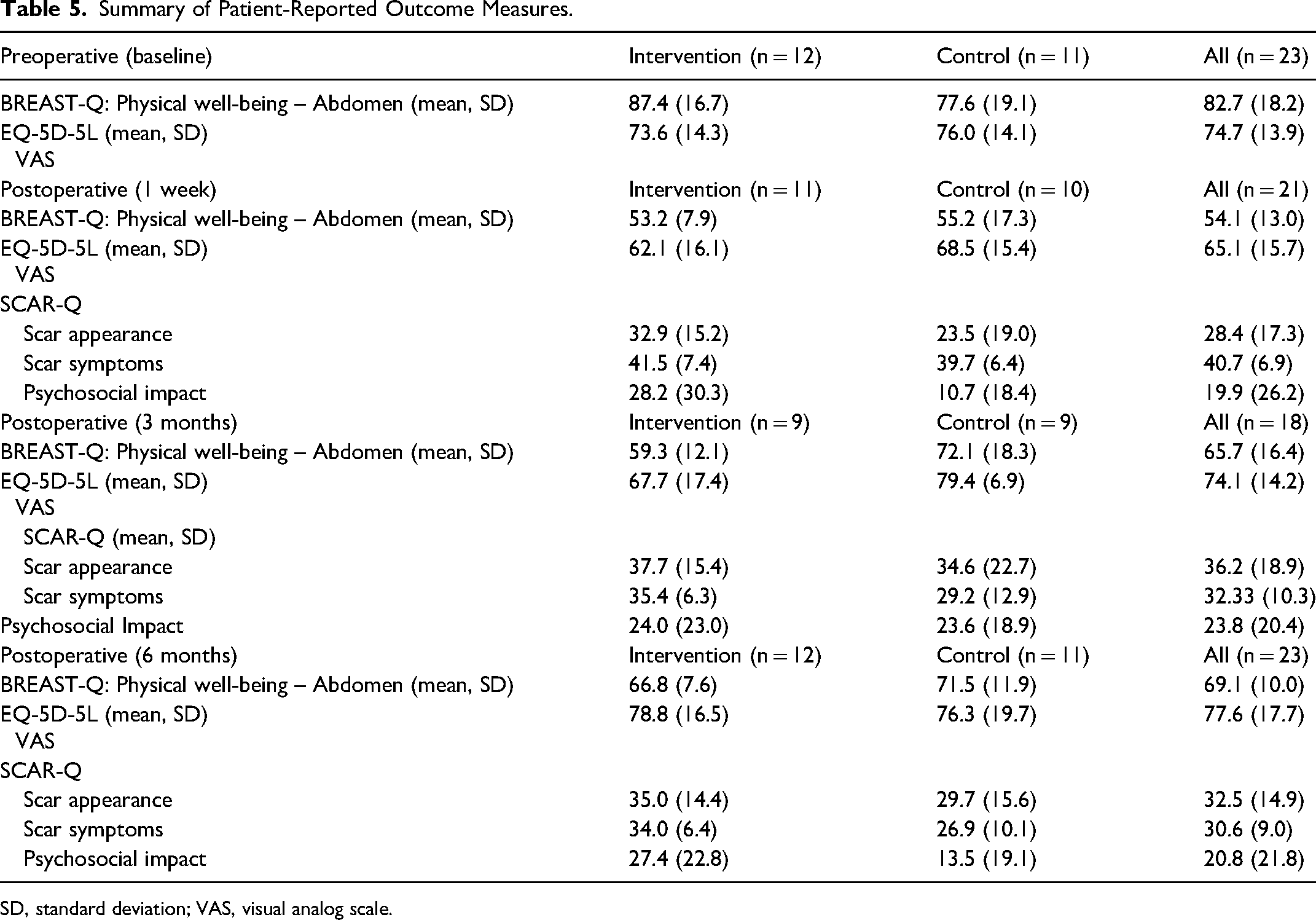
SD, standard deviation; VAS, visual analog scale.
Sample Size Calculation
Based on the wound dehiscence rates of 16.7% in the intervention and 41.7% in the control group, 51 patients will be required in each group to reject the null hypothesis that the failure rates for both groups are equal with probability (power) 0.8. The Type I error probability associated with this test of this null hypothesis is 0.05. To account for 10% loss to follow-up, we will aim to study 57 patients per group (n = 114 patients total).
Discussion
This pilot trial assessed the feasibility of conducting an RCT comparing ciNPT versus standard tape dressings for abdominal incisions in patients receiving DIEP reconstruction. Overall, the full RCT was deemed to be feasible without modifications to the protocol based on the feasibility outcomes.
There were several factors that contributed to successfully meeting all feasibility outcomes. Our eligibility rate was high (90.6%), likely due to the broad inclusion criteria. The only patient-related factors in our exclusion criteria were pregnancy, allergy to adhesives, and high BMI. In terms of recruitment, our process for recruitment was to have a member of the study team first approach the patient to introduce the study and ask if they would be interested in hearing more. Afterwards, our study coordinator would contact them to obtain consent and explain more details about the study. We found this strategy to be generally successful and contributed to our high recruitment rate (86.2%). Reasons for declining to participate included hesitancy about being randomized and already feeling overwhelmed with undergoing the surgery itself. Finally, our pre- and post-randomization rates were high, with 95.8% of patients completing the 12-month follow-up questionnaires. We were able to achieve this by consenting patients in advance to receive reminder phone calls to complete their follow-up questionnaires.
In terms of clinical outcomes, the incidence of wound dehiscence by 6 weeks was 16.7% in the intervention group and 41.7% in the control group. It is important to note that the present trial is not adequately powered to assess the effect of the intervention on clinical outcomes. The estimated sample size required for the definitive trial is 57 patients in each group, for a total of 114 patients. Notably, there was a similar RCT of 80 patients conducted by Muller-Sloof et al in the Netherlands and published in 2022, prior to the completion of this pilot trial. 21 In this trial, although the authors reported a statistically significant absolute risk reduction (ARR) of 14.5% (95% CI: 1.5-41.7). 21 However, while the ARR is easy to interpret, it is not generalizable across populations with varying risk. Given the high degree of heterogeneity of wound dehiscence rates in the DIEP population (ranging from 3.5% in the literature to 41% in Muller-Sloof et al's trial), relative risk (or risk ratio; RR) is the more valuable statistic, which was not statistically significant at 0.474 (95% CI: 0.221-1.017).21,22 Given the moderate to large effect size of the RR, an adequately powered trial may have achieved statistical significance. Thus, there is value in proceeding with the authors’ definitive RCT.
There were some challenges in the trial that resulted in minor post hoc protocol changes. Originally, the plan was to assess for abdominal site complications (ie, wound dehiscence, seroma, surgical site infection) at 2 weeks and 4 weeks. However, there were several patients who had complications just after the 4-week mark. To more accurately capture abdominal complications in the clinically relevant time period, the time was extended to 6 weeks. This change will be implemented in the definitive RCT. Additionally, we had intended to capture clinical photographs of abdominal site complications to allow for objective assessments of length, width, and depth. However, this was found to be logistically difficult and was thus removed from the protocol.
Conclusion
The authors successfully conducted a pilot RCT randomizing patients to ciNPT versus standard tape dressings for postoperative care of abdominal incisions following DIEP breast reconstruction. Based on the feasibility outcomes, the authors have decided to continue with the full RCT without modifications. The estimated required sample size will be 57 patients per group. At the time of writing, the authors have expanded the study to be a multicenter RCT and are in the recruitment process.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This study was approved by the Hamilton integrated Research Ethics Board (HiREB) (#13155) and registered on ClinicalTrials.gov (NCT04985552). All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008 (5). Informed consent was obtained from all patients for being included in the study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this research was provided by the St. Joseph's Healthcare Hamilton Studentship Award, the McMaster Surgical Associates (MSA) grant, and the Regional Medical Associates (RMA) grant.