
Abstract
Restenosis remains a significant limitation of vascular intervention procedures, primarily driven by the proliferation and inflammation of human-derived vascular smooth muscle cells (hVSMCs). This study developed a thermosensitive chitosan/β-glycerophosphate (CS-β-GP) hydrogel reinforced with graphene oxide (GO) for the localized co-delivery of paclitaxel (PTX) and dexamethasone (DEX). The incorporation of GO increased the storage modulus from approximately 3.95 kPa to 6.96 kPa, enhancing mechanical strength and accelerating gelation near body temperature (~36°C–37°C). SEM analysis revealed a porous, interconnected structure that supports drug delivery, while swelling was moderate (~145% at 6 h) and degradation resistance was improved, with the hydrogel retaining 80% of its mass in PBS after 30 days. Drug loading favored PTX (70% at 60 μg/mL) over DEX (17%) with sustained release profiles. Biologically, the dual-drug hydrogel (CS-β-GP-GO@PTX-DEX) selectively reduced hVSMC viability, migration, proliferation (~30%, ~20%, and ~30%, respectively), while preserving endothelial cell viability (~60%) and lowering proinflammatory cytokines (IL-1β ~100 pg/mL, IL-6 ~110 pg/mL, TNF-α ~70 pg/mL). These findings establish the CS-β-GP-GO@PTX-DEX hydrogel as a mechanically robust, selective, and effective platform for preventing restenosis and enhancing vascular tissue regeneration.
Introduction
Cardiovascular diseases remain the leading cause of illness and death worldwide, with vascular stenosis representing one of its most significant clinical challenges.1,2 Current interventional approaches, such as percutaneous transluminal angioplasty and stent placement, are widely used to restore blood vessel patency. However, their long-term effectiveness is often undermined by the high incidence of restenosis.3,4 The underlying pathophysiology involves mechanical disruption of the vascular endothelium during intervention, initiating a cascade of inflammatory responses. Activated endothelial and immune cells release cytokines and growth factors that stimulate the proliferation and migration of vascular smooth muscle cells (VSMCs) into the intimal layer, resulting in extracellular matrix deposition and luminal narrowing. Despite the advent of drug-eluting stents and drug-coated balloons, which release antiproliferative agents such as paclitaxel (PTX), restenosis rates remain clinically significant.5,6
PTX is particularly effective at suppressing VSMC proliferation by stabilizing microtubules and disrupting mitotic spindle formation. However, poor aqueous solubility, a narrow therapeutic index, and dose-dependent systemic toxicity hinder its clinical utility. While local delivery from stent surfaces improves therapeutic targeting, it is associated with drawbacks such as delayed endothelialisation, late stent thrombosis, and the requirement for prolonged dual antiplatelet therapy.7,8 Besides, systemic spillover from drug-coated devices can cause off-target effects. These challenges highlight the need for alternative delivery systems that achieve high local drug concentrations at the vascular injury site without the complications of permanent implants. 9 Hydrogels have emerged as promising platforms for localized therapy due to their high-water content, structural similarity to native extracellular matrix, and ability to encapsulate and release bioactive agents in a sustained manner. 10 Chitosan (CS), a cationic polysaccharide obtained via deacetylation of chitin, exhibits favorable biological properties including biodegradability, biocompatibility, antimicrobial activity, and low immunogenicity. 11 When combined with β-glycerophosphate (β-GP), CS forms a thermosensitive hydrogel that remains fluid at room temperature and undergoes sol–gel transition at physiological temperature, enabling minimally invasive administration and in situ gelation at the target site. This feature is particularly advantageous for perivascular applications, where injectable formulations can conform to the vessel surface and provide sustained drug release without a surgical procedure.12,13
Despite these advantages, conventional CS-β-GP hydrogels suffer from poor mechanical strength and limited structural stability under physiological conditions, limiting their use in dynamic environments such as the perivascular-related research. 14 Reinforcement with nanomaterials offers a viable strategy to address these shortcomings. Graphene oxide (GO), an oxidized form of graphene decorated with hydroxyl, epoxy, and carboxyl groups, combines exceptional tensile strength with high aspect ratio and abundant surface chemistry. 15 These oxygen-containing functionalities promote strong electrostatic and hydrogen-bond interactions with chitosan, while the large π-conjugated domains enable adsorption of hydrophobic drugs and biomolecules. Incorporation of GO into polymer matrices has been shown to enhance mechanical stability, thermal resistance, and protein-binding capacity, as well as to support cell adhesion, proliferation, and differentiation.16,17 In tissue engineering, GO-containing scaffolds have demonstrated the ability to modulate cellular behaviors, making them suitable for regenerative medicine applications. 17
For vascular restenosis prevention, a dual-acting therapeutic strategy that addresses both proliferative and inflammatory pathways is essential. Dexamethasone (DEX), a potent synthetic glucocorticoid, exhibits strong anti-inflammatory effects by suppressing the production and activity of inflammatory mediators, reducing matrix metalloproteinase activity, and facilitating tissue repair.18,19 Its inclusion alongside PTX can attenuate the inflammatory signaling cascade triggered by endothelial injury, which contributes significantly to neointimal hyperplasia and restenosis. Moreover, Dex’s favorable biosafety profile and established clinical use emphasize its potential as an adjunctive agent to improve vascular remodeling and reduce adverse remodeling.20,21 Co-delivery of these agents in a single platform offers the potential for synergistic suppression of both proliferative and inflammatory pathways, addressing two critical factors of restenosis. An injectable, GO-reinforced CS/β-GP hydrogel could thus oblige as a mechanically robust, thermosensitive carrier capable of sustained, localized release of PTX and Dex at the perivascular site.
In this study, we report the fabrication and in vitro evaluation of a GO-reinforced CS/β-GP thermosensitive hydrogel designed for the co-delivery of PTX and Dex. We hypothesized that the incorporation of GO would significantly enhance the mechanical and rheological properties of the hydrogel without compromising its biocompatibility, while enabling more controlled drug release profiles. The formulation was characterized in terms of physicochemical properties, gelation behavior, mechanical stability, drug release kinetics, and cytocompatibility. Our goal is to establish a multifunctional, minimally invasive drug delivery platform with the structural integrity and therapeutic efficacy required to serve as a next-generation perivascular treatment for the prevention of restenosis
Materials and methods
Materials
Medium molecular weight chitosan (molecular weight 20 kDa, deacetylation degree 85% was purchased from Titan Scientific Co., Ltd. of Beijing, China. Dissodium salt of β-Glycerophosphate, Acetic acid (AR grade) was obtained from Sigma Aldrich Co. (US). Graphene oxide in a single-layer form, with a purity greater than 99%, a lateral size range of 0.5–5 µm, and a thickness between 0.8 and 1.2 nm, was procured from XFNANO Materials Technology Co., Ltd. (Nanjing, Jiangsu, China). All subsequent solutions were prepared under continuous stirring at room temperature (25 ± 2°C) unless otherwise specified.
Synthesis of the hydrogels
A total volume of 50 mL CS-β-GP-GO hydrogel was formulated using the following procedure. First, 1 g of chitosan (CS) powder was dissolved in 30 mL of 0.75% (v/v) acetic acid and stirred vigorously for 1 h. Separately, predetermined quantities of graphene oxide (GO) nanosheets (10 mg) were dispersed in 10 mL of deionized water and sonicated for 1 h, while 3 g of β-glycerophosphate (β-GP) was dissolved in 10 mL of deionized water. The GO suspension was then combined with the CS solution and mixed continuously for 1 h to ensure homogeneity. At this stage, 25 mg of Paclitaxel (PTX) and 50 mg of Dexamethasone (DEX) were incorporated into the GO-CS mixture under gentle stirring to ensure homogeneous distribution of the drugs. Subsequently, the GP solution was added dropwise into the drug-loaded GO-CS mixture while stirring constantly until gelation occurred. For the control formulation (CS-β-GP hydrogel), the same steps were carried out, except that 10 mL of deionized water was used in place of the GO dispersion. Drug-free hydrogels were also prepared following the same protocol for comparison.
Physicochemical characterization of the hydrogels
The FTIR spectra of CS, β-GP, GO, PTX, Dex, and all the lyophilized samples were recorded in KBr pellets with a GX-1 (PerkinElmer, Waltham, MA), with a spectral range of 400–4000 cm−1, and were analyzed. The surface and internal structures of the hydrogels were examined using scanning electron microscopy (SEM). For sample preparation, hydrogel specimens were cast into Petri dishes, frozen at −80°C for 3 days, and subsequently dried by lyophilization, following a modified version of a previously reported method. 22 After drying, the samples were coated with a thin gold layer (~6.5 nm) under an argon environment at a working pressure of 4 Pa for approximately 1.3 min using a JEOL Smart Coater (Japan), and then visualized with the SEM (JSM-6010LA, JEOL, Japan).
Rheological measurement
The gelation behavior of the hydrogels was characterized using a rheometer (HAKKE Viscotester IQ Air 260-100, Thermo Fisher, USA), with approximately 2 mL of CS-β-GP, CS-β-GP-GO samples. Oscillatory rheological measurements were carried out at a frequency of 1 Hz, while the temperature was gradually increased from 20°C to 60°C at a heating rate of 1°C/min. Throughout the process, the viscoelastic parameters, namely the storage modulus (G′) and the loss modulus (G′′), were recorded under oscillatory shear conditions. The gelation point was identified as the temperature at which G′ and G′′ intersected, corresponding to a phase angle (δ) of 45°.
Mechanical properties
The compressive properties of the hydrogels were evaluated using a universal testing machine (SANS CMT4000) equipped with a 9 N load cell at ambient temperature. Sample dimensions were precisely recorded with a digital calliper prior to testing. Each specimen was subjected to compression at a crosshead speed of 2 mm/min until 20% strain was achieved. A minimum of four samples from each group were analyzed. The force–displacement data obtained during testing were converted into stress–strain curves, and the elastic modulus was calculated from the linear region of these curves.
In vitro swelling analysis
The swelling capacity of the hydrogels was determined using a standard gravimetric approach, adapted from previous reports. 23 Lyophilized hydrogel samples (n = 5) were first weighed to obtain their dry mass (W1), then immersed in distilled water at 37°C for up to 6 h. At predetermined time intervals (10, 20, 30 min, 1, 2, 4, and 6 h), the samples were removed, gently blotted with filter paper to eliminate excess surface water, and reweighed (W2). The swelling index was determined using the following formula:

Degradation profile of hydrogels
The biodegradation behavior of the hydrogels was assessed using a protocol adapted from earlier reports. 24 CS-GP-GO and CS-GP-GO@PAX-DEX formulations were incubated in 20 mL of phosphate-buffered saline (PBS, pH 7.4) supplemented with either 0 or 1000 U/mL lysozyme. CS-GP-GO and CS-GP-GO@PAX-DEX samples were maintained at 37°C under mild agitation (50 rpm) for 30 days. At specific time intervals, the hydrogels were collected, rinsed with PBS to remove residual enzymes or buffer, freeze-dried, and subsequently weighed. The extent of degradation was expressed as the percentage of weight loss, calculated according to the following equation
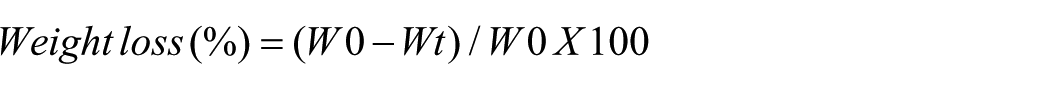
where W0 is the initial dry weight of the sample and Wt is the dry weight at time t.
Evaluation of drug release
The release behavior of Paclitaxel (PAX) and Dexamethasone (DEX) from CS-GP-GO@PAX-DEX hydrogels was evaluated in phosphate-buffered saline (PBS), pH 7.4. For each test, 10 mg of the drug-loaded hydrogel was dispersed in 10 mL of PBS and incubated at 37°C under continuous shaking in glass tubes. At predetermined time intervals, 0.5 mL of the release medium was withdrawn and immediately replaced with an equal volume of fresh PBS to maintain sink conditions. Drug concentrations in the collected samples were quantified using high-performance liquid chromatography (HPLC) with an external standard method. 25 The cumulative drug release percentage was determined according to the following equation:

where Mt represents the amount of drug released at time t, and M0 is the total drug content initially loaded into the hydrogel.
Encapsulation efficiency and drug loading efficiency of CS-GP-GO@PAX-DEX
The encapsulation efficiency and drug loading of Paclitaxel (PAX) and Dexamethasone (DEX) were determined following a procedure adapted from the previous literature. 26 Briefly, varying concentrations of PAX or DEX (1, 2.5, 5, 10, 30, 50, and 60 µg/mL) were dissolved in ethanol. To each solution, 10 mg of CS-GP-GO was introduced and subjected to sonication for 2 min, and the pH was adjusted to 3 using HCl. The mixtures were stirred at 4°C in the dark for 24 h, then neutralized to pH 7.8 with NaOH and stirred for another hour. The resulting suspensions were centrifuged at 10,000 rpm for 20 min, and the precipitates were washed twice with deionized water and freeze-dried overnight and designated as CS-GP-GO@PAX and CS-GP-GO@DEX, respectively.
For quantification, 10 mg of each drug-loaded hydrogel was dissolved in 1 mL of ethanol, vortexed (3 min), and sonicated (15 min) to ensure complete disintegration of the nanocarriers. The concentrations of PAX and DEX were determined using a UV/Vis spectrophotometer (Shimadzu UV1800, Japan) at wavelengths of 227 and 240 nm, respectively, with calibration curves as references. The encapsulation efficiency (EE) and drug loading content (DLC) of CS-GP-GO@PAX and CS-GP-GO@DEX were calculated using the following equations:
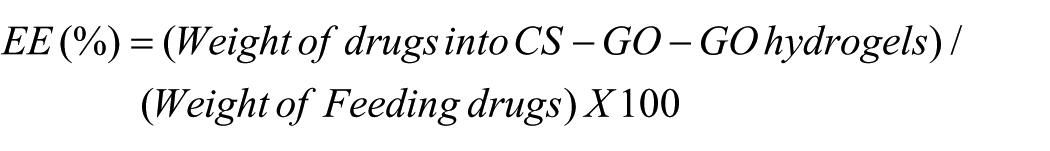

Injectability and injection force evaluation
The injectability of the CS-β-GP-GO hydrogel was evaluated by measuring the injection force using a universal mechanical testing machine (Instron 5943, Instron Corporation, Shanghai, China) equipped with a calibrated load cell. The hydrogel was loaded into a disposable syringe connected with the needles with different gauges (18G, 25G, and 27G) and lengths (25G × 1½″ and 25G × ¼″), while maintaining the volume of hydrogel constant. The sample-filled syringers were safely mounted on the base platform of the testing machine. The load cell was carefully aligned to ensure that it contacted the syringe plunger only after force calibration. Injection was performed in tensile extension mode at a fixed crosshead speed, allowing the hydrogel to be extruded in a controlled and reproducible manner. The injection force was continuously recorded for 1–2 min. The effects of needle gauge and needle length on injectability were evaluated by comparing the peak force and the subsequent steady plateau force, with the corresponding force–time curves used to assess the stability and uniformity of hydrogel flow during injection. All experiments were carried out at a flow rate of 2 mL/h.
Cell culture
Human-derived ECs and VSMCs were procured from iCell Bioscience Inc. (Shanghai, China). Cultures were kept at 37°C under 5% CO₂ using Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum plus 1% penicillin/streptomycin.
All hydrogel-related experiments, CS-GP-GO hydrogel with and without PTX and DEX, were synthesized under sterile conditions as per the details given above and underwent sol-gel transition at 37°C before cell exposure. Unless otherwise mentioned, hydrogel samples were utilized in the pre-gelled state to ensure consistent contact with cells.
Live and dead cells assay
To evaluate the cytocompatibility, the pre-gelled hydrogels such as CS-βGP-GO, CS-βGP-GO@PTX, CS-βGP-GO@DEX, and CS-βGP-GO@PTX-DEX were placed in 24-well plates (200 μL per well). hVSMCs and hECs were seeded directly on the surface of hydrogles at a density of 1 × 104 cells per well. The seeded cells were cultured with PBS (control), CS-GP-GO, and its drug-loaded derivatives (CS-GP-GO@PTX, CS-GP-GO@DEX, and CS-GP-GO@PTX-DEX) for 5 days. Cell viability was subsequently assessed using a Live/Dead Viability Kit (Thermo Fisher Scientific, USA) following the manufacturer’s instructions. Briefly, cells were incubated with Calcein-AM and propidium iodide (PI) for 30 min at 37°C and examined using confocal laser scanning microscopy (Olympus CKX53, Tokyo, Japan). Viable cells emitted green fluorescence due to intracellular esterase-mediated hydrolysis of Calcein-AM, a nonfluorescent, membrane-permeable probe. In contrast, nonviable cells exhibited red fluorescence as PI, a membrane-impermeant dye, which selectively intercalated with nuclear DNA.
Migration assay
hVSMCs and hECs were cultured in 6-well plates to reach 90%–95% confluence (n = 3 per group). A linear wound was generated by scraping the monolayer with a sterile 200 μL pipette tip. The plates were gently washed with PBS to remove detached cells. Then the cells were incubated with the serum-free culture medium containing CS-βGP-GO, CS-βGP-GO@PTX, CS-βGP-GO@DEX, or CS-βGP-GO@PTX-DEX hydrogels. The pre-gelled hydrogel samples (10 mg/mL) were incubated in serum-free DMEM at 37°C for 24 h to obtain hydrogel conditioned media. This media were then added to the scratched cells monolayer and incubated for 24 h at 37°C in a 5% CO₂ atmosphere. The cell migration across the wound area was visualized under an optical microscope (Olympus CKX53, Japan). The wound closure distance was quantified using ImageJ software, and the migration rate (%) was determined according to the equation:

where A0 represents the initial wound width and At represents the wound width at the indicated time point.
Cell proliferation assay
BrdU immunocytochemistry was conducted to evaluate the proliferation of hVSMC and hEC. Briefly, 1 × 10⁵ cells were seeded in 96-well plates and allowed to adhere overnight under standard conditions. The culture medium was replaced with the hydrogels conditioned media (given in the earlier section) and the cells were incubated for additional 24 h. BrdU (25 μM) (Cat. B5002; Sigma-Aldrich, St. Louis, USA) was added to each well and incubated for 16 h. Cells were then washed, fixed with 4% paraformaldehyde for 20 min at 4°C, and subjected to 1 N HCl for 15 min at 37°C to denature DNA. For immunostaining, cells were incubated with anti-BrdU primary antibody (Cat. B8434; 1:1000; Sigma-Aldrich) prepared in PBS containing 0.1% Triton X-100 and 2.5% BSA for 2 h at room temperature. After three PBS washes, antigen–antibody complexes were visualized using an Alexa Fluor-488–conjugated secondary antibody, and the sample was incubated for 1 h at 37°C. Stained cells were imaged with a fluorescence microscope (Olympus CKX53, Japan). Proliferation was quantified as the ratio of BrdU⁺ cells to the total number of cells in each treatment group. Statistical analysis was performed using an unpaired t-test (GraphPad Prism v8.0).
Assessment of pro-inflammatory cytokines by ELISA
Vascular smooth muscle cells (hVSMCs) and endothelial cells (hECs) were individually seeded into 96-well plates at a density of 1 × 104 cells per well and incubated overnight at 37°C in a humidified atmosphere containing 5% CO₂ to allow cell adhesion. To induce oxidative stress, cells were treated with 20 μL of hydrogen peroxide (H₂O₂) at a final concentration of 300 μM for 2 h. Following this, cells were washed with PBS and incubated with hydrogel conditioned media prepared from CS-βGP-GO, CS-βGP-GO@PTX, CS-βGP-GO@DEX, or CS-βGP-GO@PTX-DEX formulations. While PBS treated medium served as the control. Cells were further incubated under standard conditions for up to 24 h. Cell culture supernatants were collected at 24 h post-treatment, centrifuged at 1000 × g for 10 min to remove cellular debris, and analyzed for pro-inflammatory cytokines, tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6). Quantification was performed using sandwich ELISA kits (eBioscience, USA) according to the manufacturer’s protocols.
Results and discussion
Physicochemical characterization
The schematic representation of the synthesis of CS-βGP-GO@PTX-DEX from the monomer units is depicted in Figure 1(a). The FT-IR spectrum provides clear evidence of the molecular interactions among chitosan (CS), β-glycerophosphate (β-GP), and their complex (CS-β-GP), as well as with GO, PTX, and DEX, through distinct band shifts and changes in transmittance as given in Figure 1(b). For chitosan, broad peaks around 3478 cm⁻1 are attributed to O–H and N–H stretching, which are characteristic of both the polysaccharide backbone and the amino functionalities. The presence of bands near 2901 cm⁻1 corresponds to C–H stretching vibrations, affirming the presence of methyl and methylene groups. Amide I (C=O stretching) and amide II (N–H bending) appear at approximately 1657 cm⁻1, indicating the presence of residual acetyl groups and active amine groups from chitosan. β-GP exhibits broad O–H stretching about 3341 cm⁻1 and significant P–O stretching between 1074 cm⁻1, confirming phosphate and hydroxyl functional groups. 27 The CS-β-GP complex spectrum shows reductions and shifts in the O–H and N–H stretching region, indicating the formation of hydrogen bonds and possible ionic interactions between the CS and β-GP molecules. The emergence and intensification of peaks related to P–O stretching in the complex, as well as the appearance of new bands attributed to C=O–NH (amide bond formation) near 1659 cm⁻1, further support the complexation and the establishment of strong intermolecular forces. Additional absorbances, such as those associated with PO₄³⁻ (969 cm⁻1) and possible NO₂ groups (around 770 cm⁻1), confirm the formation of specific interactions that result in a structurally integrated biomaterial.14,27 The FT-IR spectrum of graphene oxide clearly indicates that the material contains key oxygen-based functional groups. The broad peak at 3357 cm⁻1, corresponding to O-H stretching, indicates the presence of hydroxyl groups. Peaks at 2909 cm⁻1 and 1603 cm⁻1 confirm the presence of C-H and C=O bonds. Other notable signals at 1478, 1266, and 1154 cm⁻1 show vibrational signatures of water, epoxy, and carboxyl groups. The FT-IR spectrum of the CS-β-GP-GO composite confirms successful integration of all components. A broad band at ~3300 cm⁻1 (O–H/N–H) and peaks near 2901 cm⁻1 (C–H) reflect chitosan’s hydroxyl, amino, and aliphatic groups. Amide I/II signals at 1667 cm⁻1 indicate C=O/N–H interactions from CS–βGP bonding.14,27 Strong P–O stretches between 1130 cm⁻1 reveal β-GP’s phosphate linkages. Shifts and intensity changes across these regions demonstrate hydrogen bonding, ionic interactions, and amide formation, evidencing a structurally cohesive composite suitable for biomedical use.

(a) Schematic representation of the synthesis of CS-β-GP-GO@PTX-DEX hydrogel, (b) FT-IR spectrum of CS, β-GP, CS-β-GP, GO, CS, β-GP-GO, PTX, DEX, and CS, β-GP-GO@PTX-DEX, SEM images of (c) CS-β-GP, (d) CS-β-GP-GO, (e) CS-β-GP-GO@PTX-DEX.
The FT-IR spectra further provided clear evidence of distinct functional groups in bare PTX, DEX, and their dual-incorporated composite CS-β-GP-GO@PTX-DEX. For PTX, a broad absorption at approximately 3430 cm⁻1 signifies O–H and N–H stretching, characteristic of the hydroxyl and amide functionalities in its structure. A strong peak near 1735 cm⁻1 corresponds to ester carbonyl (C=O) stretching, while peaks in the 1595–1510 cm⁻1 region arise from aromatic C=C and amide II vibrations. Additional features observed between 1250 and 1050 cm⁻1 represent C–O–C stretch, supporting the presence of ester and ether groups within PTX. 28 DEX exhibits similar but distinct signals: the O–H stretching peak is observed around 3380 cm⁻1, reflecting the abundant hydroxyl groups, and a carbonyl stretching band appears near 1662 cm⁻1. Notable fingerprint peaks between 1200 and 1000 cm⁻1 can be assigned to C–O stretching typical of DEX’s ring structure and ether linkages. 29 In the composite CS-β-GP-GO@PTX-DEX spectrum, the above key peaks are present but often shifted or broadened, indicating successful incorporation and molecular interactions between PTX, DEX, and the hydrogel matrix. The broad O–H/N–H region around 3400 cm⁻1 and a carbonyl signal approximately between 1700 and 1650 cm⁻1 suggest overlapping contributions from both drugs and matrix. New or intensified features near 1200–1000 cm⁻1 reflect composite bonding changes, confirming effective drug integration. 30 Collectively, the spectral modifications and the presence of these specific functional groups reinforce that the composite forms a complex network through multiple intermolecular forces, including hydrogen bonding, ionic interactions, and covalent linkages.
The SEM images reveal the microstructural evolution of hydrogel composites through successive modifications. The CS-β-GP hydrogel (Figure 1(c)) exhibits a highly porous, three-dimensional network characterized by abundant interconnected pores, a hallmark of efficient hydrogel formation that supports both high water uptake and molecular transport. 30 When graphene oxide is introduced to make the CS-β-GP-GO hydrogel (Figure 1(d)), the morphology changes distinctly, the structure appears more fibrous, and the pore walls thicken, suggesting reinforced mechanical strength while retaining significant porosity. This transformation emphasizes GO’s function as a structural enhancer within the hydrogel matrix, giving rise to a more robust yet permeable framework. 14 Upon subsequent loading with PTX and DEX (CS-β-GP-GO@PTX-DEX, Figure 1(e)), the micrographs reveal that the composite maintains its porous network, though the pore size distribution becomes more uniform and the surface smoother, indicative of successful drug encapsulation. 31 These alterations likely result from drug molecules occupying some of the internal cavities and further facilitating physical crosslinking. The combination of preserved interconnected porosity and increased morphological order following drug loading reflects the hydrogel’s potential for sustained drug release and biocompatibility. 31
Rheological characterization and gelation dynamics of hydrogels
The rheological profiles presented in Figure 2(a) to (d) provide a quantitative perspective on thermally-induced and time-dependent gelation kinetics for the CS-β-GP and CS-β-GP-GO hydrogels. Figure 2(a), corresponding to CS-β-GP, shows a progressive increase in storage modulus (G′), starting below 1 Pa at 20°C and rising sharply beyond 60 Pa as the temperature approaches 60°C, signifying rapid network formation and transition from liquid to gel. The loss modulus (G″) remains consistently lower, peaking at just above 10 Pa, which reflects that elastic characteristics predominate in the final gel state, ensuring structural integrity. 14 For CS-β-GP-GO hydrogel (Figure 2(b)), G′ values similarly climb from below 1 Pa to above 70 Pa over the same temperature range, with the final modulus slightly outperforming the unmodified hydrogel. This enhancement suggests that graphene oxide incorporation confers additional stiffness and crosslinking, promoting more resilient gel formation. The G″ remains well below G′, maintaining a maximum around 12 Pa, so the hydrogel exhibits pronounced elastic dominance.14,32 The intercept points observed in Figure 2(a) and (b) represent the gelation temperature (T_gel) for the CS-β-GP and CS-β-GP-GO hydrogels, respectively. This point occurs where the storage modulus (G′) crosses over the loss modulus (G″), marking a critical transition in the material’s behavior from predominantly viscous (liquid-like) to predominantly elastic (solid-like).33,34 In Figure 2(a), this crossover is noted around 37.4°C, indicating that the CS-β-GP hydrogel begins to form a stable gel above this temperature. Similarly, in Figure 2(b), the crossover points shift slightly to a lower temperature near 36.2°C for CS-β-GP-GO, suggesting that the inclusion of graphene oxide facilitates earlier gelation.14,32 This shift implies enhanced intermolecular interactions or additional crosslinking sites introduced by GO, enabling more rapid transition into the elastic gel state.14,32
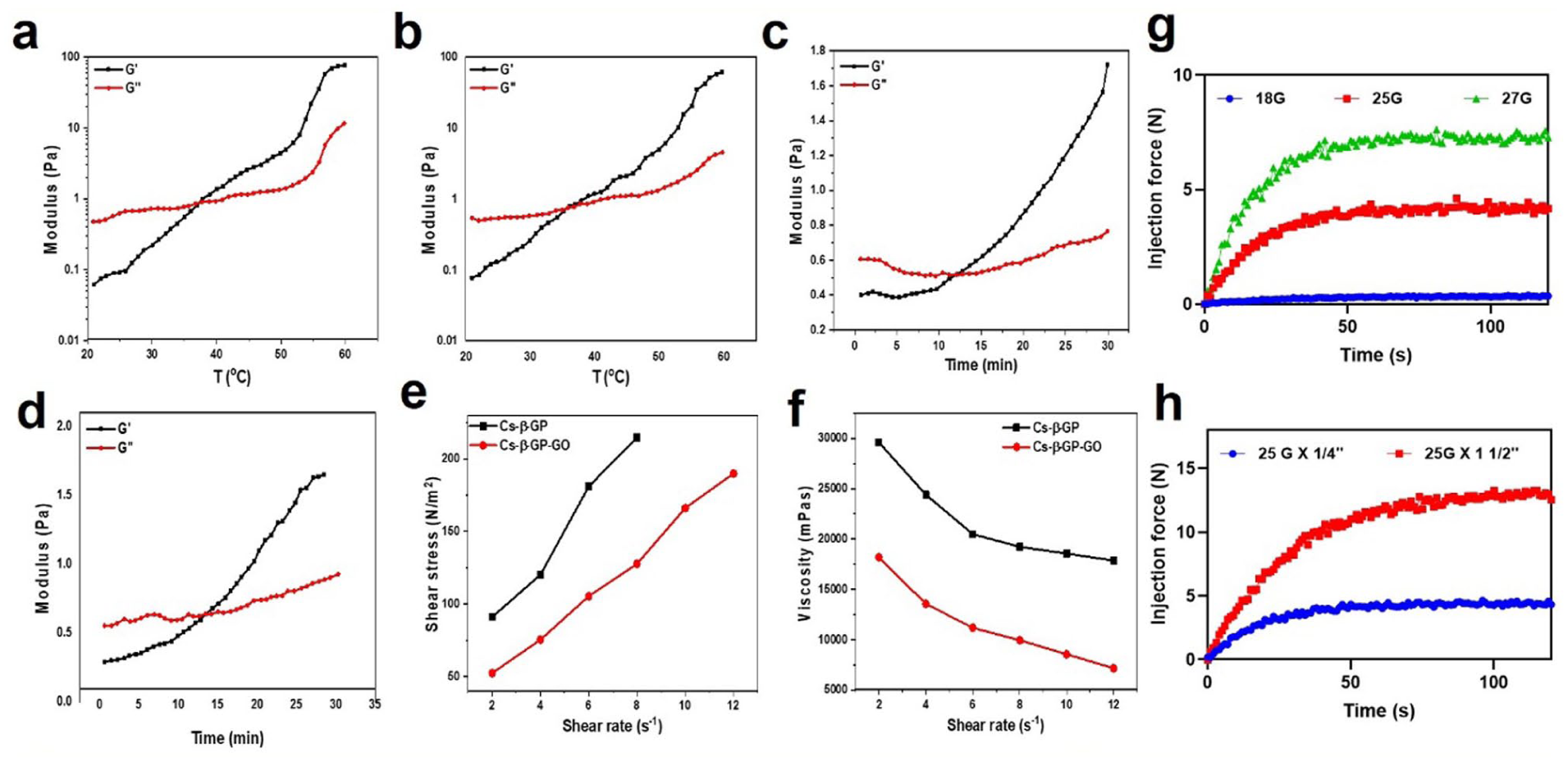
(a, b) Storage modulus of CS-β-GP, CS-β-GP-GO as a function of temperature (20°C–60°C), (c, d) storage modulus of CS-β-GP, CS-β-GP-GO as a function of time at the temperature 37°C, (e) the relationship between shear stress and shear rate of CS-β-GP, CS-β-GP-GO measured at 37°C, (f) the influence of shear rate on the rheological curves of CS-β-GP, CS-β-GP-GO measured at 37°C, (g) graph of injection force over time for CS-β-GP-GO injected from a 1 mL syringe with 1/4 in. with the needle different gauges (18G, 25G, and 27G), (h) and the 1 mL syringe with a 25G needle of 1 1/2 in. and 1/4 in. length.
Figure 2(c) and (d) depict the time sweep data under isothermal conditions for each material. CS-β-GP (Figure 2(c)) exhibits a G′ increase from 0.4 Pa at the initial time point to around 1.6 Pa at 30 min, outpacing G″ (which fluctuates gently around 0.6 Pa).14,32 In contrast, CS-β-GP-GO (Figure 2(d)) starts at roughly 0.5 Pa and quickly rises to 1.8 Pa by 30 min. The elevated G′ values relative to G″ throughout signify successful gel maturation and improved viscoelastic properties, with GO conferring measurable improvement in network strength and stability.33,34 Overall, these results confirm rapid and robust gelation for both hydrogel systems, with graphene oxide enhancement leading to superior mechanical properties. The elastic character (G′ > G″) across all conditions ensures that both hydrogels can maintain shape, support cell growth, and resist deformation, making them optimal for biomedical applications requiring in situ gelation and resilience characteristics essential for injectable scaffolds and drug delivery platforms in high-impact therapeutic research.34,35 In Figure 2(c) and (d), the intercept point where the storage modulus (G′) crosses over the loss modulus (G″) represents the gelation time (t_gel) of the hydrogels at a constant temperature. This point marks the transition from a predominantly viscous liquid state to a predominantly elastic gel state, indicating the onset of solid-like behavior in the material. For CS-β-GP (Figure 2(c)), the crossover occurs at approximately 8 min, signifying that the hydrogel begins to gel and form a stable network around this time. In contrast, CS-β-GP-GO (Figure 2(d)) shows the intercept point earlier, at about 6 min, demonstrating that the incorporation of graphene oxide accelerates the gelation process.35–37 The shorter gelation time in the GO-containing hydrogel suggests enhanced crosslinking kinetics, likely due to increased interactions between polymer chains and GO nanosheets, which facilitate faster network formation.35–37 This accelerated gelation is beneficial for practical applications where rapid in situ solidification is desirable, such as injectable biomaterials for drug delivery or tissue engineering, ensuring quicker stabilization at the target site while maintaining desirable mechanical properties.36–38
The rheological behavior depicted in Figure 2(e) and (f) offers important insights into the flow characteristics of CS-β-GP and CS-β-GP-GO hydrogels. In Figure 2(e), shear stress increases steadily with rising shear rate for both formulations, with CS-β-GP consistently exhibiting higher values compared to CS-β-GP-GO. For example, at a shear rate of 2 s⁻1, CS-β-GP shows a shear stress around 100 N/m2, which escalates to more than 200 N/m2 at 12 s⁻1. In contrast, CS-β-GP-GO begins at approximately 60 N/m2 and reaches 180 N/m2 at the highest shear rate. This trend demonstrates that incorporating graphene oxide reduces the resistance to deformation, indicating easier flow under applied stress.38,39 Figure 2(f) reveals distinct shear-thinning characteristics in both hydrogels, a symbol of non-Newtonian fluids. The viscosity of CS-β-GP starts above 28,000 mPas at 2 s⁻1 and drops to just below 18,000 mPas at 12 s⁻1. CS-β-GP-GO displays even lower initial viscosity, around 18,000 mPas at 2 s⁻1, decreasing to about 8000 mPas at the highest rate. These results affirm that GO incorporation not only decreases both shear stress and viscosity but also enhances flowability without compromising structural properties.38,39 The improved shear-thinning behavior of CS-β-GP-GO is particularly advantageous for applications requiring minimally invasive administration and uniform distribution, supporting its candidacy for advanced biomedical uses as injectable hydrogels or drug delivery matrices.37–39
The injectability test was performed using a mechanical testing machine to determine the injection force of the CS-β-GP-GO hydrogel. The injection force profile as a function of time shows a clear difference associated with the needle gauge and length (Figure 2(g) and (h)). In all cases, the injection force increased rapidly during the initial phase, then slowly reached a plateau. Indicating the establishment of a stable flow of hydrogel through the needle. As shown in Figure 2(g), injection force increased with the decrease in the needle diameter. The smaller 27G needle required highest force (~7 N), followed by the 25G needle (~4 N), and the broader 18G shown the minimal resistance (~0.4 N) throughout the injection. This profile infers that the flow resistance was increased, associated with the smaller needle diameter. The graph for all the needles shows a smooth and stable curve, showing the flow of the hydrogel is uniform without any clogging or disturbance. Further, the effect of needle length on the injection force was obtained with 25G × 1½″ and 25G × ¼″ as given in Figure 2(h). The longer 25G × 1½″ needle required a higher injection force (~12 N) than the smaller needle (~4 N) consistently, due to the increased frictional resistance and pressure drop associated with a longer flow path. Despite this increase, both needle lengths reached steady plateau forces without notable fluctuations, and disturbances representing the uniform flow behavior of hydrogel. Overall, the gradual force buildup and stable plateaus confirms the shear-thinning behavior of hydrogel. This make the hydrogel easy to inject under stress and show resistance in the absence of stress. This characteristic is favorable for the minimally invasive delivery, and the injectability could be turned by choosing the appropriate needle selection without compromising the flow, making this hydrogel system suitable for injectable biomedical applications.24,32,35,40
Swelling behavior and degradation kinetics of hydrogels
The swelling and degradation profiles highlight clear differences in performance between the CS-β-GP and CS-β-GP-GO hydrogels, offering crucial insights into their suitability for biomedical applications. As shown in the swelling experiments (Figure 3(a)), both hydrogels exhibit an initial rapid uptake of water, consistent with the highly porous structures observed previously. At the initial time point (0 h), CS-β-GP shows a swelling rate of approximately 240%, while CS-β-GP-GO starts lower at 215%. Over the next 6 h, swelling rates reduce and stabilize: at 6 h, CS-β-GP maintains approximately 165%, whereas CS-β-GP-GO holds near 145%. This consistent reduction in swelling for CS-β-GP-GO can be attributed to the presence of GO within the polymer network, which likely introduces additional cross-linking and density, thereby restricting water absorption while simultaneously enhancing the mechanical stability of the matrix.14,41,42
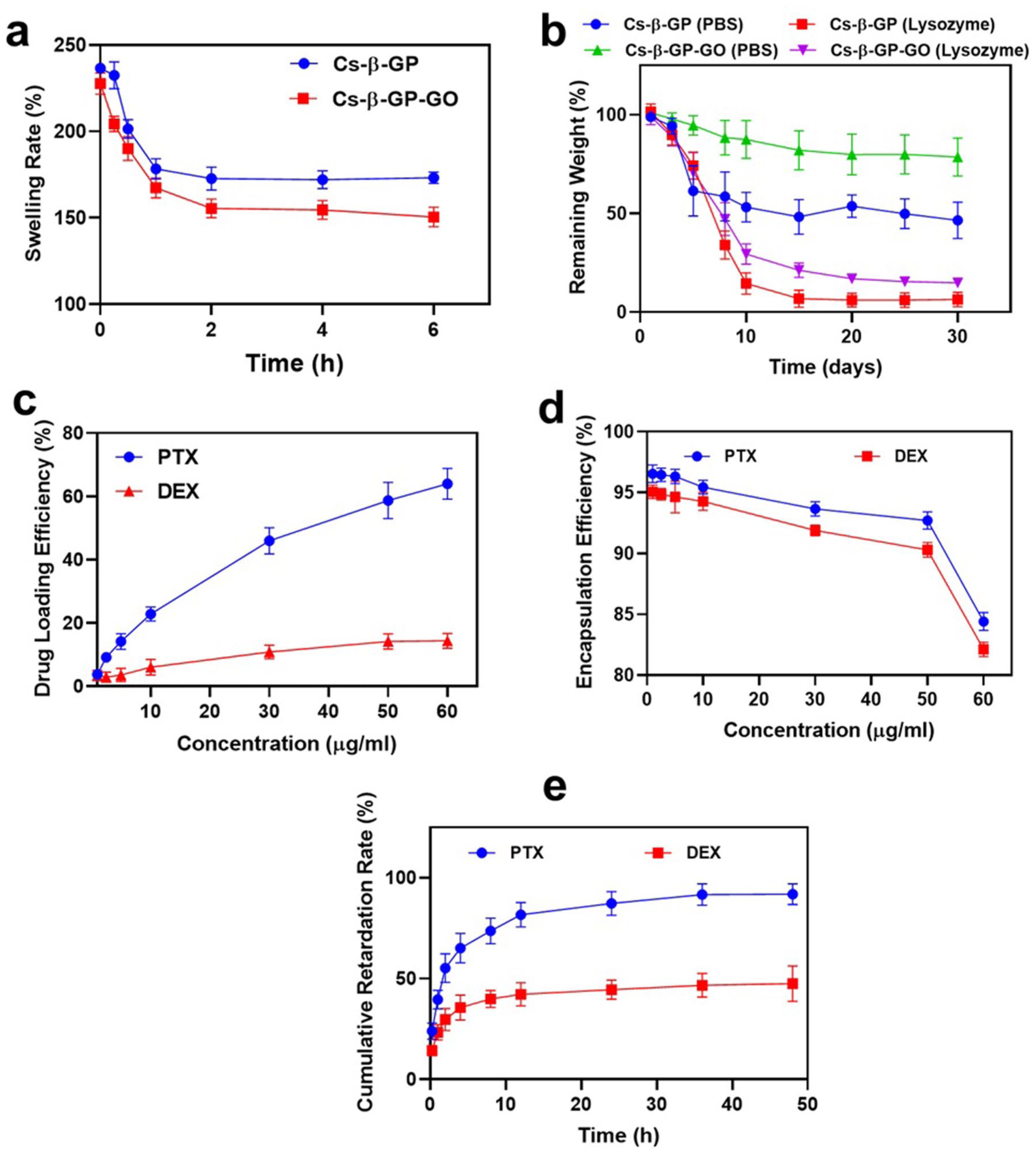
(a) Swelling ratio of CS-β-GP, CS-β-GP-GO hydrogels in distilled water at 37°C for up to 6 h, (b) degradation studies of CS-β-GP, CS-β-GP-GO hydrogels in the presence of PBS, and lysozyme for 30 days, (c) drug loading efficiency of PTX, DEX in CS-β-GP-GO, (d) encapsulation efficiency of PTX, DEX in CS-β-GP-GO, (e) in vitro release of PTX, and DEX from CS-β-GP-GO matrix.
Degradation studies further emphasize the structural advantages imparted by GO. Over a 30-day period (Figure 3(b)), CS-β-GP hydrogel displays a marked decrease in remaining weight, particularly in the presence of lysozyme, indicating rapid enzymatic breakdown.14,43 In contrast, CS-β-GP-GO retains a substantially greater proportion of its initial mass: in PBS, roughly 80% remains after 30 days, and in lysozyme, it maintains about 20%. Both hydrogels degrade more quickly in lysozyme than in PBS. This enhanced resistance to degradation is a reflection of GO’s reinforcing effect, providing greater stability against hydrolytic and enzymatic attack.14,41,43 Notably, while both hydrogels degrade more quickly in lysozyme than in PBS, the GO-integrated formulation’s superior durability positions it as a promising material for applications requiring prolonged residence time and sustained release, such as drug delivery implants or tissue engineering scaffolds.43–45
Drug loading efficiency, encapsulation, and release profiles in CS-β-GP-GO hydrogels
The presented data delineate the nuanced interactions between the CS-β-GP-GO hydrogel matrix and two drugs, PTX and DEX, across drug loading, encapsulation efficiency, and release kinetics, offering a comprehensive overview of the system’s drug delivery potential. The drug loading efficiency chart (Figure 3(c)) illustrates a clear difference between the two molecules: PTX, loading efficiency rises from approximately 15% at 5 μg/mL to about 70% at 60 μg/mL, indicating strong and steadily increasing affinity for the hydrogel network. 46 In contrast, DEX shows much lower uptake, starting near 5% and only reaching around 17% at maximal concentration, suggesting limited interaction with the matrix even as concentration increases. 47 This discrepancy may be attributed to variance in molecular size, hydrophobicity, and affinity for the GO-containing polymer network, indicating that the hydrogel matrix possesses a greater binding capacity for PTX. 48
Regarding encapsulation efficiency (Figure 3(d)), both drugs start with high values above 95% at 5 μg/mL, emphasizing the hydrogel’s proficiency in entrapping therapeutic agents. As concentration grows, encapsulation efficiency for PTX drops moderately to 85% at 60 μg/mL, whereas DEX declines more sharply, falling to approximately 80% at the same concentration. This pattern indicates more robust retention mechanisms for PTX and highlights a tendency for DEX to saturate or escape the hydrogel matrix as concentration increases.49–51
The cumulative release profile (Figure 3(e)) provides further insight. PTX exhibits an initial burst release, rapidly reaching over 65% within the first 5 h, and continues to increase to nearly 95% by 50 h. DEX, however, shows a gradual release, with the retardation rate only progressing from about 30% at 5 h to 50% at 50 h, pointing to stronger retention or slower diffusion through the hydrogel structure.50–52 Such controlled release dynamics, rapid for PTX, protracted for DEX, are highly desirable for tailoring treatments where sequential or sustained therapeutic delivery is necessary.51,52 Together, these results convey a synergistic formulation capable of both high encapsulation and tailored release, reinforcing the hydrogel’s application in precision drug delivery where distinct pharmacokinetic requirements must be balanced for optimal therapeutic outcomes.
In vitro analysis
Live and dead analysis
Assessment of cell viability using live/dead staining provides meaningful differences in biological response between vascular smooth muscle cells (hVSMC) and endothelial cells (hEC) exposed to various hydrogel formulations (Figure 4(a)). On Day 1, both cell types show predominantly green fluorescence across all samples, indicative of high viability and good initial biocompatibility. By Day 5, however, the addition of DEX to the PTX hydrogel formulation (CS-β-GP-GO@PTX-DEX) produces a pronounced increase in red-stained (live cells 30%), nonviable hVSMCs compared to single-drug or control samples (live cells 90%), indicating that dual loading significantly enhances cytotoxic effects on these cells. Quantitative analysis reveals a significant drop in the percentage of live hVSMC cells, from ~90% in the control to ~30% with the dual-drug hydrogel, ~85% with CS-β-GP-GO alone, ~50% with PTX, and ~65% with DEX-loaded samples.53–56 This robust reduction in live hVSMCs falling below 40%, while single-drug and control formulations remain above 85%–90% demonstrates that dual drug loading significantly potentiates cytotoxic action against smooth muscle cells. Such suppression aligns with desired outcomes in vascular therapy, where limiting hVSMC proliferation is critical to prevent restenosis.53–56
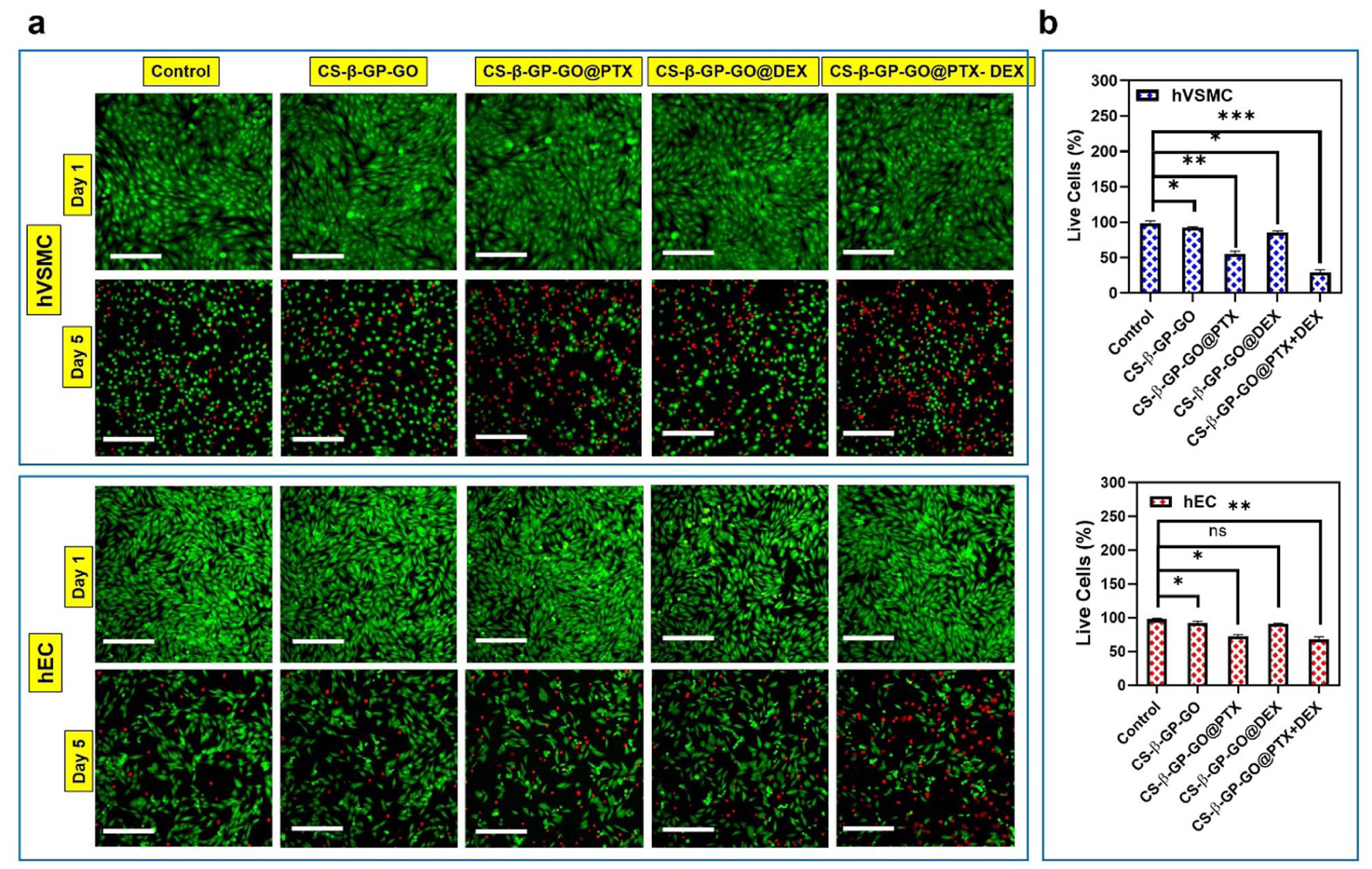
(a) Fluorescence micrographs stained by Calcein-AM/PI of hVSMC and hEC with 25 μg/mL of PBS, CS-β-GP-GO, CS-β-GP-GO@PTX, CS-β-GP-GO@DEX, and CS-β-GP-GO@PTX-DEX, (b) quantitative results of the live cell % obtained from the micrographs for hVSMC and hEC, respectively.
In contrast, hEC viability follows a more moderate trajectory, with the most substantial reduction seen in dual-drug samples but overall survival remaining higher than in hVSMCs. On Day 5, the dual-drug system (CS-β-GP-GO@PTX-DEX) reduced viability to roughly ~60%, whereas controls and single-drug hydrogels sustained about ~95% and ~80% (PTX) and ~85% (DEX), respectively.57,58 While single-agent hydrogels exhibit more moderate effects, hEC viability in the dual-drug group shows a substantial yet less severe decrease than hVSMC, suggesting selective cytotoxicity is achievable for these formulations, maximizing inhibition of smooth muscle while limiting endothelial cell loss. Together, these results highlight the promise of drug-laden hydrogels, especially dual-loaded systems, for precise cell targeting and improved efficacy in vascular therapy.53–58
Wound closure assay
The wound closure assay clearly demonstrates the pronounced effects of the various hydrogel formulations on cellular migration in both human vascular smooth muscle cells (hVSMC) and endothelial cells (hEC) (Figure 5). Microscopic images taken at 0 and 24 h (Figure 5(a)) indicate rapid closure of the wound gap in the control and CS-β-GP-GO groups for both cell types, confirming strong migratory capacity in the absence of drugs. By comparison, samples loaded with PTX, DEX, and particularly the dual-drug CS-β-GP-GO@PTX-DEX system show considerably slower gap closure, with visible open areas persisting even after 24 h, especially in the hVSMC monolayers.55,56 Quantitative analysis (Figure 5(b)) shows the control group achieves about 85% migration closure, while CS-β-GP-GO reaches approximately 75% closure at 24 h. When exposed to drugs, the closure rate drops: CS-β-GP-GO@PTX and CS-β-GP-GO@DEX demonstrate intermediate suppression, with closure values near 50% and 55%, respectively.58,59 The strongest inhibitory effect is observed in the CS-β-GP-GO@PTX-DEX group, where migration closure falls to just ~20%, highlighting profound restraint on hVSMC movement. This drastic reduction in migration closure in the dual-drug group, dropping below 20% compared to 85% in controls, demonstrates a potent capacity for curbing unwanted smooth muscle migration, a key therapeutic goal for post-angioplasty restenosis prevention.58–60

(a) Representative images from the wound closure assay of hVSMC and hEC treated with PBS, CS-β-GP-GO, CS-β-GP-GO@PTX, CS-β-GP-GO@DEX, and CS-β-GP-GO@PTX-DEX for 24 h, (b) bar graph showing the percentage of migration closure of various drugs treated at 24 h in the wound closure assay.
For endothelial cells (hEC), migration rates remain relatively higher than in hVSMCs, even under drug treatment, indicating partial preservation of wound-healing ability. The control group closes around 90% of the migration gap after 24 h, while the CS-β-GP-GO group is close behind at ~80%. Drug-loaded hydrogels exert greater inhibition: CS-β-GP-GO@PTX and CS-β-GP-GO@DEX reduce migration closure to approximately 65% and 60%, while the CS-β-GP-GO@PTX-DEX results in closure of about 40%, lower than single-drug systems but still allowing partial endothelial repair.55–58 These results suggest the dual drug system is especially potent against smooth muscle cells while preserving some endothelial regenerative capacity. Taken together, the data highlight the enhanced efficacy of dual drug loading, with CS-β-GP-GO@PTX-DEX outperforming single-drug and control groups in limiting cellular migration.58–60 These results offer compelling evidence that a multifunctional approach combining PTX with DEX in a GO-reinforced hydrogel matrix effectively curtails hVSMC proliferation and migration, an essential feature for vascular therapies aimed at restenosis prevention and tissue integration.
Anti-proliferation assay
The BrdU proliferation assay provides valuable insight into how different hydrogel formulations influence cell division in both hVSMC and hEC. As seen in Figure 6(a), the cells treated with control medium and CS-β-GP-GO hydrogel conditioned medium consistently display strong BrdU-positive staining, indicating elevated levels of proliferating cells. In contrast, all drug-loaded hydrogels, particularly the dual drug CS-β-GP-GO@PTX-DEX formulation, show a pronounced reduction in BrdU incorporation for hVSMC, reflected by visibly fewer fluorescent nuclei.55–58 This suppression is especially evident in the combination group, suggesting an enhanced antiproliferative effect when PTX and DEX are co-delivered. Quantitative analysis (Figure 6(b)) normalized to the control group set at 100%, indicates that hVSMC proliferation remains high in the control condition. CS-β-GP-GO alone showed proliferation slightly less (~90%) than the control.55–60 Conditioned medium derived from dual-drug-loaded hydrogel produced a dramatic reduction, with CS-β-GP-GO@PTX and CS-β-GP-GO@DEX registering at approximately 40% and 55% proliferation, respectively. Most notably, the CS-β-GP-GO@PTX-DEX dual-drug hydrogel further suppresses hVSMC proliferation to just ~30%, representing the strongest anti-proliferative impact among all test groups. This striking decrease of hVSMC proliferation from ~100% in controls to ~30% in the CS-β-GP-GO@PTX-DEX demonstrates the enhanced potential for restenosis prevention, with compounded suppression observed only when PTX and DEX are co-delivered.55–61

(a) The cell proliferation assay using BrdU on hVSMC and hEC treated with PBS, CS-β-GP-GO, CS-β-GP-GO@PTX, CS-β-GP-GO@DEX, and CS-β-GP-GO@PTX-DEX, (b) quantification of BrdU proliferation % of hVSMC and EC, (c, d, e) anti-inflammatory assays of cytokines IL-1β, IL-6, and TNF-α were conducted using ELISA after 24 h of treatment with PBS, CS-β-GP-GO, CS-β-GP-GO@PTX, CS-β-GP-GO@DEX, and CS-β-GP-GO@PTX-DEX.
Endothelial cells respond slightly differently. Although their proliferation is reduced in the presence of drug-loaded hydrogels, the suppression is less dramatic than in hVSMCs, especially with the dual-drug system.58–60 BrdU analysis reports control hEC proliferation at about 95%, with CS-β-GP-GO hydrogels eliciting a small decline to ~90%. PTX and DEX single treatments lower hEC proliferation to ~70% and ~80%, respectively. The CS-β-GP-GO@PTX-DEX hydrogel moderately reduces hEC proliferation, settling near 65%, thus retaining a substantial repair and regeneration capacity compared to hVSMCs.55–61 This selective inhibition is promising, as it preserves the capacity for endothelial repair and regeneration crucial for vessel healing post-intervention while simultaneously limiting pathological smooth muscle expansion.
Synergistic anti-inflammatory impact of dual drug hydrogels
Analysis of the ELISA results for proinflammatory cytokines IL-1β, IL-6, and TNF-α across both hVSMC and hEC reveals compelling evidence of the modulatory effects of hydrogel-conditioned media derived from the given formulations (Figure 6(c)–(e)). Control and CS-β-GP-GO groups record the highest cytokine levels across all panels, with IL-1β at around 200 pg/mL, IL-6 near 280 pg/mL, and TNF-α close to 150 pg/mL. The introduction of single-drug hydrogels leads to significant declines; PTX lowers IL-1β, IL-6, and TNF-α to approximately 160, 185, and 120 pg/mL, while DEX brings these values further down to about 145, 170, and 95 pg/mL, respectively.58–61 Most notably, cells treated with conditioned media derived from CS-β-GP-GO@PTX-DEX dual-drug hydrogel display the lowest cytokine concentrations, roughly 100 pg/mL for IL-1β, 110 pg/mL for IL-6, and 70 pg/mL for TNF-α—showcasing potent, synergistic anti-inflammatory effects. This descending trend with combined PTX and DEX treatment underscores the synergistic anti-inflammatory effect of dual loading.55–61 Notably, hEC cytokine levels are consistently lower than those measured in hVSMCs, suggesting a comparatively reduced inflammatory response in endothelial cells, which may be beneficial for vascular healing. The magnitude of cytokine reduction is especially pronounced for IL-6 and TNF-α, both critical mediators in vascular remodeling and pathological restenosis.57–61 These results conceptually align with earlier observations from BrdU proliferation and wound closure assays, confirming that dual drug hydrogels not only restrain unwanted cell growth and migration but also minimize the local inflammatory milieu. Emphasizing the benefit of dual drug delivery, the marked decrease in all proinflammatory cytokines supports the strategy’s therapeutic relevance for advanced vascular biomaterials. By reducing inflammation more effectively than either drug alone, the CS-β-GP-GO@PTX-DEX hydrogel provides comprehensive control over post-implantation cellular responses, potentially curbing complications and accelerating tissue integration. Collectively, these findings validate the dual-drug approach’s superiority in mitigating both hyperproliferation and inflammation, making it an ideal candidate for clinical translation in vascular regenerative medicine. However, PTX and DEX in the CS-β-GP-GO hydrogel effectively control restenosis by reducing hVSMC proliferation (~70%), migration (~75%), and inflammation; they may delay endothelial healing or wound closure if overdosed. Though hECs retain ~60% to 65% viability/migration, prolonged exposure could disrupt vessel repair post-angioplasty. Future work must optimize dosing, kinetics, and peri-adventitial delivery to balance pathology control with regeneration. Although the in vitro studies demonstrated favorable selective cytotoxic, anti-proliferative, and anti-inflammatory responses of the CS-β-GP-GO@PTX-DEX hydrogel, the in vivo behavior of GO nanosheets requires further investigation. In particular, their long-term fate, systemic distribution, clearance, and potential tissue accumulation remain unclear. Comprehensive animal studies are therefore necessary to evaluate biocompatibility, safety, and translational relevance, including considerations related to nanomaterial toxicity and regulatory approval.
Conclusion
The CS-β-GP-GO hydrogel system, incorporating PTX and DEX, presents a compelling approach for vascular regenerative medicine by combining mechanical robustness, biocompatibility, and controlled drug delivery. Strong intermolecular bonding within the composite forms a stable matrix with a highly porous structure that supports sustained release. The inclusion of graphene oxide enhances gel stiffness and accelerates gelation near body temperature, facilitating injectable applications. The hydrogel also demonstrates improved swelling control and degradation resistance, ensuring longer functional durability. Drug loading favors PTX with effective encapsulation, while drug release profiles enable a balanced therapeutic effect. Importantly, the dual-drug hydrogel selectively suppresses pathological vascular smooth muscle cell behaviors such as viability, migration, and proliferation. While preserving endothelial cell function, supporting vascular healing. It also effectively reduces inflammatory cytokines, contributing to a favorable microenvironment for tissue regeneration. Together, these results establish the CS-β-GP-GO@PTX-DEX hydrogel as a promising multifunctional platform with the potential to prevent restenosis and enhance vascular repair, offering significant clinical relevance for advanced vascular therapies.
Footnotes
Author contributions
Jinping Sheng: Conceptualization, investigation, data curation. Rui Jiang: Methodology, investigation, writing – original draft. Peng Wang: Methodology, investigation, writing – original draft. Jianhao Li: Data curation, writing – review and editing. Feizhou Du: Supervision, validation, conceptualization, writing – review and editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Management Project of the General Hospital of the Western Theater Command (No. 41C416D5).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All additional data from this study will be made available on request.*
Statement on the use of generative AI
The authors utilized Grammarly solely for language editing and grammatical correction, and Perplexity as an aid in identifying pertinent sources and conducting a comprehensive literature review. All material was independently reviewed, modified where necessary, and approved by the authors, who take full responsibility for the reliability and integrity of the final manuscript.