
Abstract
Background:
Congenital bladder disorders in children necessitate innovative approaches for bladder tissue regeneration, aiming to minimize complications associated with conventional therapies. This study focused on generating a cell-seeded scaffold using superior smooth muscle cells (SMCs) by exploring the potential of smooth muscle cell spheroids (3D SMCs) compared to conventionally cultured SMCs (2D SMCs) for bladder tissue engineering. Additionally, adipose-derived stem cells (ADSCs) were investigated for their impact on SMC proliferation and maturation, and pre-differentiated smooth muscle-like ADSCs (pADSCs) for their potential as alternative cell source.
Methods:
3D SMCs were seeded into a compressed collagen scaffold as monoculture and as co-culture with ADSCs or pADSCs and incubated for 2 weeks. Their contractile potential as well as proliferation and cell distribution within the scaffold were compared to conventionally cultured 2D SMCs by immunofluorescent staining and qRT-PCR.
Results:
3D SMCs in collagen scaffolds exhibited significantly superior cell distribution, proliferation, and contractile marker expression compared to 2D SMCs. While ADSCs showed limited impact, co-culture with pADSCs enhanced contractile marker expression, though not surpassing 3D SMC monoculture.
Conclusion:
For the first time, a collagen scaffold seeded with 3D SMCs was generated and evaluated. This study recommends 3D SMCs as optimal building blocks for bladder tissue engineering, highlighting the potential of pADSCs as an alternative cell source. These findings offer crucial insights for refining cell sources as well as culture techniques in pediatric bladder regeneration and provide a superior cell-seeded scaffold for further bladder tissue engineering experiments.
Introduction
Congenital disorders such as spinal dysraphism, posterior urethral valves, or bladder exstrophy can cause bladder wall fibrosis, function loss, and early-age renal damage in children. 1 Primary therapy involves anticholinergic medications and clean intermittent catheterization. If unsuccessful, surgical bladder augmentation with an intestinal segment is necessary to prevent kidney damage.2,3 However, the incorporation of intestinal tissue into the bladder may lead to severe complications, significantly reducing the patient’s quality of life.2–4 Tissue engineering (TE) is an alternative approach that aims to reduce morbidity due to the use of the intestinal segment. The regeneration of urologic tissue derived from the patient’s own cells represents an attractive option, particularly for pediatric patients with a distinct need for a long-lasting and functional tissue substitute that grows with the patient. 5 Unfortunately, early tissue fibrosis and lack of functionality have hindered successful clinical applications to date.6,7
Challenges in bladder TE include identifying suitable biomaterials, enhancing cell integration and functionality, and ensuring the survival of the implanted scaffold in vivo.5,8 Regarding the biomaterial, three main classes are currently in use: acellular tissue matrices, including bladder submucosa and small-intestinal submucosa; synthetic polymers, including polyglycolic acid, polylactic acid, and polylactic-co-glycolic acid; and naturally derived materials, such as alginate and collagen. 8 Collagen type I, in particular, has demonstrated an excellent microenvironment for cell seeding.9,10 Additionally, its biomechanical stability can be enhanced through compression. 11 Our group recently tested a cell-seeded compressed collagen hydrogel for bladder TE, proving its feasibility and regenerative capabilities in vivo. 12 Regarding the functionality of the regenerated bladder tissue, elasticity and contractility are crucial for the bladder’s regulated filling and voiding. Elasticity is primarily determined by the composition of the extracellular matrix, which consists mainly of collagen and elastin.13–15 The contractile potential is determined by the smooth muscle cells (SMCs) of the bladder wall. Unfortunately, de-differentiation of SMCs from a contractile to a synthetic phenotype during in vitro culturing, leading to fibrosis of the implanted scaffold, is a known issue in bladder TE. 5 Even SMCs isolated from healthy rat urinary bladders demonstrated a reduced contractile potential after multiple passages of conventional cell culture expansion. 16 The most commonly applied strategies to maintain the cell phenotype include adaption of cell culture microenvironment, employment of growth factors, optimization of biomaterial properties, and mechanical stimulation. 5 Using three-dimensional (3D) cell cultures, or so-called spheroids, is another strategy to address the low contractility. This method offers more natural cell clusters with superior properties, at least ex vivo, compared to conventional SMC cultures. 5 Spheroids are spherical cell aggregates of primary cell monocultures or a co-culture of different cell lines cultured as free-floating units. 17 They mimic embryonic development, during which cells interact closely with each other.18,19 Spheroid formation relies on the cells’ ability to produce their own extracellular matrix and does not require exogenous biomaterials such as preformed biodegradable scaffolds. 19 Cells within spheroids express improved regenerative properties, strong angiogenic and vasculogenic capacity, and higher growth factor production than two-dimensional (2D) cultures. 20 Unlike organoids, spheroids lack the ability to self-organize into complex structures through cellular differentiation. However, their simplicity offers advantages regarding culture time, and cost.21,22 Their use has been established in cancer and drug delivery research and is of growing interest in the field of regenerative medicine.23,24 As previously shown, the utilization of 3D cell cultures with SMCs (3D SMCs) isolated from rodent detrusor muscles is feasible and results in SMCs that express a greater quantity of functionally relevant contractile markers, heightened differentiation, and improved contractile potential when compared to conventionally cultured SMCs (2D SMCs). 25 Additionally, creating 3D SMCs from diseased human detrusor tissues restores a contractile phenotype closer to the healthy state. 26
Another strategy to restore the contractile phenotype of SMCs is the use of autologous stem cells with the potential to transform diseased into healthy cells.27,28 Of the various stem cell types assessed, multilineage adipose-derived stem cells (ADSCs) have been the focus of interest for bladder TE. 5 They can be easily harvested from the subcutaneous tissue and have been proposed as a highly proliferative and multipotent cell line to support SMC remodeling and tissue formation by secreting growth factors, cytokines, and extracellular matrix proteins.5,27–31 ADSCs, especially when cultured as spheroids, produce higher growth factors and improve cell survival in ischemic tissue. 32 Pre-differentiating ADSCs into smooth muscle-like pADSCs, expressing SMC-specific markers, provides an alternative autologous cell source for diseased bladder tissue.12,31,33 This is of importance since, especially in children with impaired bladders, the cultivation of sufficient quantities of healthy SMCs represents a further challenge in bladder TE.
Bladder spheroid cultures from urothelial or cancer cell lines are already in use as in vitro models for cancer research as well as treatment studies for other urologic diseases such as stress urinary incontinence and urinary tract infections.23,34 Little attention has been paid to the use of spheroids for bladder TE applications in terms of generating a cell-seeded scaffold ready for implantation. Bladder TE requires a dense, interconnected cell distribution throughout the scaffold. However, up to date, it has not been investigated whether 3D SMCs maintain their superior qualities over conventionally cultured SMCs after being seeded into a scaffold. Challenges to making spheroid cell culture a gold standard for in vivo applications include successful scaffold colonization, establishing co-cultures with supporting cell lines, and standardization of high-throughput SMC spheroid culture techniques. Spheroid production and handling are relatively straightforward, and if these spheroids retain their properties in vivo and enhance tissue regeneration, they could become a new gold-standard cell source for bladder TE, with the potential for adaptations such as co-cultivation.
Our aim in this study was to generate a cell-seeded scaffold containing functional SMCs, presenting a tissue-engineered bladder prototype, which can be used for further in vivo trials. We used the previously validated compressed collagen hydrogel as a biomaterial and combined the use of spheroid culture and adding autologous stem cells to address the low contractility in cultured SMCs as well as the low cell number. We investigated whether 3D SMCs continue to be superior building blocks for bladder TE compared to 2D SMCs, hypothesizing that 3D SMCs in compressed collagen form a denser network with higher expression of contractile markers than 2D SMCs, thus maintaining their superiority after being implemented into a biomatrix and having populated the scaffold. We also evaluated ADSCs and pADSCs as supporting cell sources. We hypothesized that ADSCs, when co-cultured with SMCs on a spheroid level, enhance SMC proliferation and maturation and that a spheroid co-culture of pADSCs and SMCs shows an equally high contractile marker expression as a 3D SMC monoculture.
Materials and methods
SMC isolation and culture
Urinary bladders were harvested from euthanized male Wistar rats following the ethical guidelines for the use of animals in research (license no. 110/2015, Ethics Committee for Animal Experimentation, ECAE). SMCs were isolated following established procedures. 25 Bladder specimens were cut open, and the urothelial layer was removed using a scalpel. The remaining detrusor muscle was minced into 1 mm pieces and air-dried for 5 min in 10 cm cell culture Petri dishes. SMC culture medium consisting of DMEM/F12 + GlutaMAX (Invitrogen, Waltham, MA, United States), 10% fetal bovine serum (FBS; Merck, Darmstadt, Germany), 1% PS, 0.5 ng/ml human basic fibroblast growth factor (hbFGF; Sigma, St. Louis, MI, USA), 5 ng/ml human epidermal growth factor (hEGF; Sigma, St. Louis, MI, USA) and 5 μg/ml human insulin (Sigma, St. Louis, MI, USA) was added, and culture dishes were incubated at 37°C with 5% CO2. The medium was changed every 3 days, and the remaining tissue pieces were removed after 1 week. SMCs were used up to passage six.
ADSC isolation and culture
To isolate ADSCs, standard protocols 35 were followed. Subcutaneous fat tissue from inguinal regions of rats was minced and digested in 0.1% (w/v) collagenase Type I (Merck, Darmstadt, Germany) for 1 h at 37°C and followed by centrifugation. The cells were resuspended in PBS and filtered through a (100 μm) filter. The pellet was resuspended in medium (DMEM/F12, Gibco, GI, USA), supplemented with 10% FBS and 1% PS. Cells were incubated at 37°C with 5% CO2. The culture medium was changed twice a week until 90% ADSC confluence was reached. ADSCs from passages 2–5 were used.
Predifferentiation of ADSCs into pADSCs
ADSCs were pre-differentiated using an established protocol. 33 Briefly, ADSCs were expanded to 80% confluence and then incubated for 3 weeks in SMC-inductive MCDB-131 medium (Merck, Darmstadt, Germany) supplemented with 1% FBS, 1% PS, and 100 U/ml heparin (Bichsel, Interlaken, Switzerland).
Cell characterization
Expanded cells were characterized in passage three before their use for further experiments by fluorescence-activated cell sorting (FACS) with lineage-specific markers. For ADSC anti-rat CD29, CD73, CD90.1, and CD45 were used. The differentiation of ADSCs into SMCs was confirmed with the SMC-related proteins anti-calponin, anti-smoothelin, anti-MyH-11and anti-α-SMA. The same SMC markers were used for the characterization of bladder-derived SMCs. The samples were detected by flow cytometry (FACS, Becton Dickinson FACS Canto flow cytometer), and data was analyzed using FlowJo software v. 7.5 (Tree Star Inc., Ashland, OR, USA). Details of antibodies are provided in Table 1. A viability count was performed for each cell culture before their use for further experiments, and cells were only used if viability was positive for more than 95% of the cells.
Details of antibodies used in experiments.
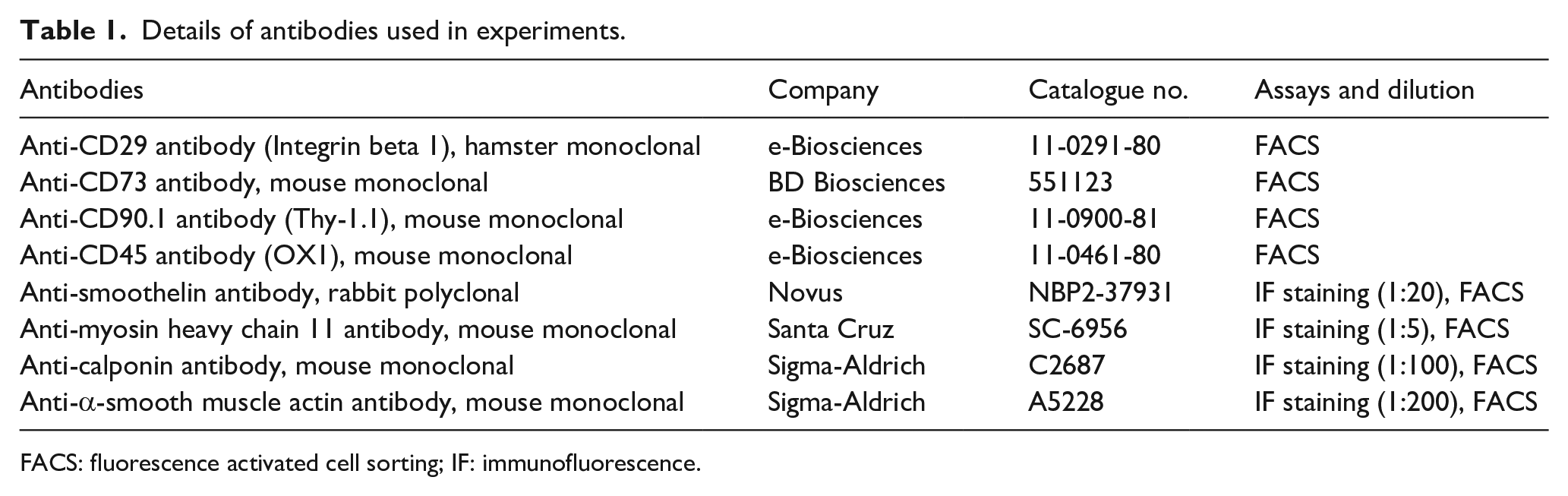
FACS: fluorescence activated cell sorting; IF: immunofluorescence.
Spheroid production
Spheroids were cultured on Sphericalplates 5D® (Kugelmeiers, Erlenbach, Switzerland) according to the manufacturer’s protocol and as previously described. 25 One plate consists of 24 wells, half of which contain 750 punched-out nonadhesive microwells. The microwells have an inverted pyramidal shape, which allows SMCs to develop close cell-cell interactions as they sediment to the bottom of the wells and form spheroids. Two milliliter of culture medium containing either a monoculture of SMCs or a 1:1 co-culture of either ADSCs/SMCs or pADSCs/SMCs were seeded per well, which equals 1500 cells per microwell or spheroid, respectively. SMC culture medium was used for every condition. Spheroids were cultured for 2 days. Characteristics, as well as concentration profiles of SMC spheroids, have been previously analyzed and published by our group. 25 Figure 1(a) and (b) shows schematic spheroid production using the different cell lines.
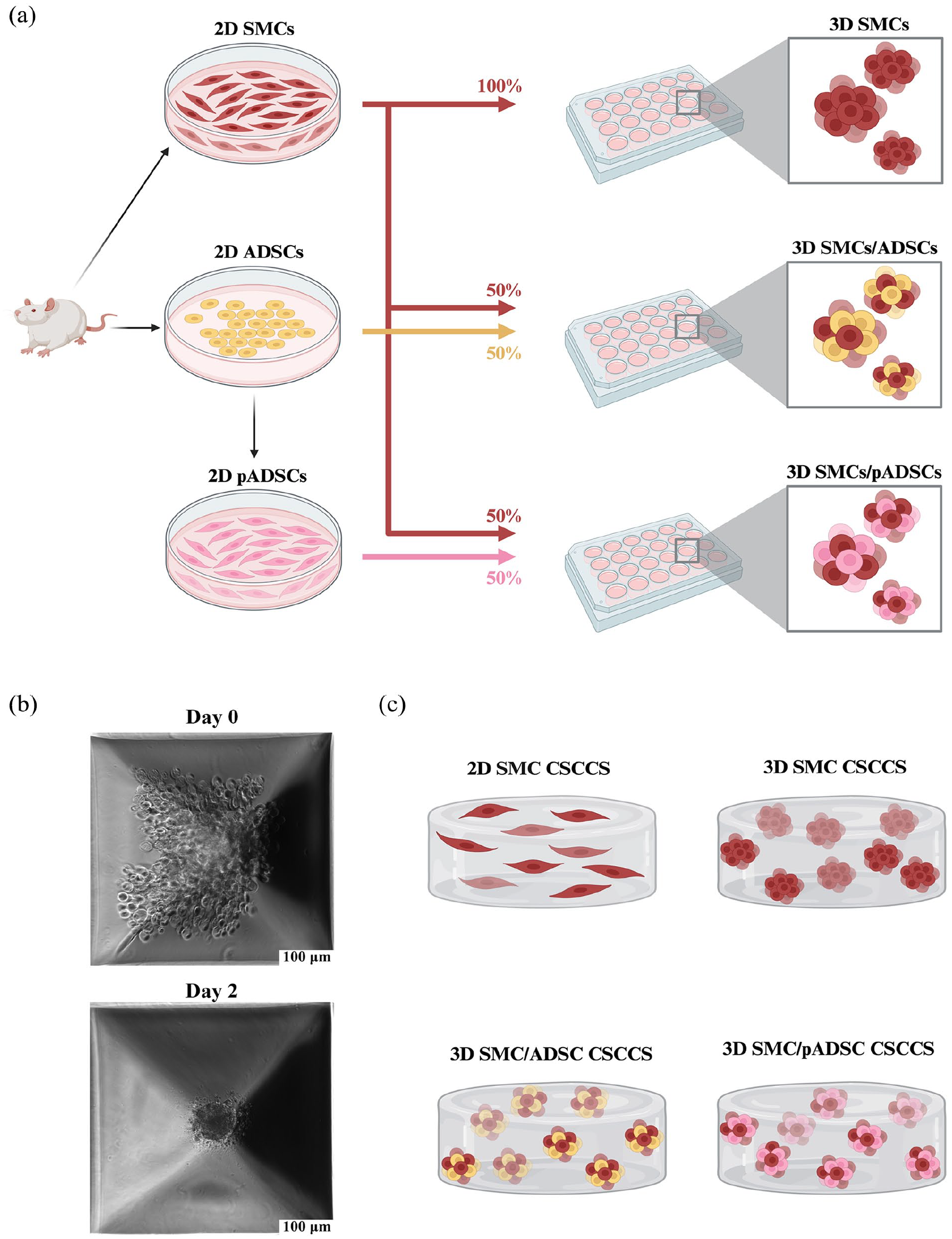
Schematic spheroid production and CSCCS setup. (a) SMCs and ADSCs were isolated from harvested urinary bladder from euthanized male Wistar rats and cultured. ADSCs were pre-differentiated into pADSCs using SMC-inductive MCDB-131 medium. Spheroids of the cultured cells were then produced by seeding solely SMCs or a 1:1 co-culture of either ADSCs/SMCs or pADSCs/SMCs into the Sphericalplates 5D®. Created in BioRender. Mazumdar, A. (2024) https://BioRender.com/g61j628. (b) Image acquisition by inverted microscope of the microwells within the Sphericalplates 5D® shows spheroid formation over 2 days. (c) Schematic single cell and spheroid setup within the CSCCS. Created in BioRender. Mazumdar, A. (2024) https://BioRender.com/e47m696.
Cell-seeded compressed collagen scaffold
The preparation of cell-seeded compressed collagen scaffold (CSCCS) was carried out according to a published protocol. 12 Briefly, the CSCCS were produced using a mixture of 1.8 ml bovine collagen type I (6 mg/ml, Advanced Biomatrix, Carlsbad, USA), mixed with 450 μl 0.189 M acetic acid (Sigma, St. Louis, USA), and 875 μl sterile filtered polymerization buffer containing 0.32 M NaHCO3 (Sigma, St. Louis, USA), 0.15 M NaOH (Merck, Darmstadt, Germany), and 0.2 M HEPES (Sigma, St. Louis, USA) in double-distilled H2O. 400 μl culture medium was added containing (a) 4.5 × 106 2D SMCs, (b) 4.5 × 106 SMCs (equivalent to 3000 SMC spheroids), (c) 2.25 × 106 SMCs + 2.25 × 106 ADSCs (equivalent to 3000 SMC/ADSC spheroids) or d) 2.25 × 106 SMCs + 2.25 × 106 (equivalent to 3000 SMC/pADSC spheroids). Figure 1(c) shows the schematic cell setup of the four CSCCS conditions. The number of cells seeded per scaffold was determined according to previously published experiments by our group. 12 The mixture was transferred to a cell culture insert with a PET track-etched membrane with a diameter of 23.1 mm and a pore size of 3.0 μm (Falcon, Corning, USA) and placed in a 6-well cell culture plate (Falcon, Corning, USA). The CSCCS were incubated for 30 min at room temperature (RT) and then 2 h at 37°C for polymerization.
To create a mechanically strong tissue, the CSCCS were compressed according to published protocols.11,12,36 The compression device previously described by our group 12 and weighing 150 g was placed on the scaffolds for 5 min, leading to a final thickness of 500 μm in the wet state, representing a 94% volume loss.11,12 To prevent CSCCS coiling, a membrane from a cell culture insert was placed over each CSCCS and weighed down with a surgical stainless-steel ring (24 mm, 0.875 g). The CSCCS were incubated for 2 weeks at 37°C with 5% CO2 in SMC culture medium in a humidified incubator before being harvested. The medium was changed every 3 days.
To confirm the mechanical stability of the harvested CSCCS, the scaffolds were cut and sutured back together. Further mechanical and functional testing of the CSCCS was dispensed with since their mechanical properties, as well as their feasibility for in vivo trials, have been previously investigated.11,12
Immunofluorescent staining of CSCCS
To investigate the contractile SMC proteins, CSCCS were stained after 2 weeks of incubation and imaged by inverted microscopy (Leica THUNDER Imaging System, Wetzlar Germany).
CSCCS samples were fixed in 4% PFA at 4°C overnight and washed in PBS. Samples were blocked with 10% BSA in PBS with 1% NGS and 0.2% Triton X-100 for 2 h. Following, primary antibodies for contractile SMC proteins anti-calponin, anti-smoothelin, and anti-MyH-11 were added, and samples were stored at 4°C overnight. Samples were washed for 4 h in PBS, the Cy3-conjugated secondary antibody was added, and samples were stored at 4°C overnight. After a washing cycle with PBS, samples were incubated for 10 min at RT with DAPI and washed in PBS for 1 h. Image acquisition was made using an inverted microscope (Leica THUNDER Imaging System, Wetzlar Germany). Exposure times were 75 ms for DAPI and 80 ms for calponin, smoothelin, and MYH-11. For negative controls, the primary antibody was omitted. Details of primary antibodies are provided in Table 1.
Cell count and assessment of cell-scaffold interaction
To assess cell density and distribution within the scaffolds, overview images of the whole CSCCS slice were acquired for each condition (tile scan). A series of images taken at a set interval between the first and last plane of focus (Z-stack) was obtained for each tile scan.
Z-stacks at 5× magnification were acquired in triplicates from DAPI-stained nuclei for each sample. A maximum intensity projection was used for each Z-stack to highlight the DAPI signal for better segmentation. Nuclei were segmented by thresholding the DAPI signal, and cell counts were obtained by using the “analyze particles” function in ImageJ. 37
Contractile protein quantification of CSCCS
For analysis of contractile SMC phenotype, proteins calponin, smoothelin, and MyH-11 were quantified by mean fluorescent intensity (MFI) divided by cell number across the camera field of view (FOV) according to a published protocol. 38 Leica Application Suite (LAS) was used for image acquisition with a 20× objective. Z-stacks were collected using the same settings to analyze different samples (Exposure: 50 ms, Fluorescent Intensity Manager: 100%). For each different primary antibody, three Z-stacks were taken in representative areas. A maximum intensity projection was used for each Z-stack and exported as a RAW file.
ImageJ® was used for quantification of fluorescent intensity. To exclude background signal, the average of three regions of interest (ROI) devoid of cells was subtracted from the MFI. Background-corrected MFIs were divided by cell number. Normalized MFI was compared between scaffolds with 2D SMCs, 3D SMCs, 3D SMCs/ADSCs, and 3D SMCs/pADSCs.
Total RNA isolation and quantitative real-time PCR
For the analysis of SMC-specific contractile gene induction within the CSCCS and the native rat bladder, qRT-PCR was conducted. Total RNA was isolated according to the manufacturer’s protocol using TRI Reagent Solution (Thermo Fisher, Waltham, MA, USA). 2 μg RNA was reverse-transcribed to cDNA with random primers (High-Capacity cDNA reverse transcription, Applied Biosystems). We evaluated the SMC-specific contractile marker genes for calponin, smoothelin, and MyH-11. The samples (50 ng of cDNA per sample) were analyzed by measuring the cycle threshold (CT) values; CT values were between 20 and 30 in all samples. Each sample was measured in triplicate and normalized to the housekeeping gene GAPDH. Fold increase in gene expression was compared between scaffolds with 2D SMCs, 3D SMCs, 3D SMCs/ADSCs, and 3D SMCs/pADSCs. Details of the primers used are provided in Table 2.
qRT-PCR primers for TaqMan gene expression assay (FAM).
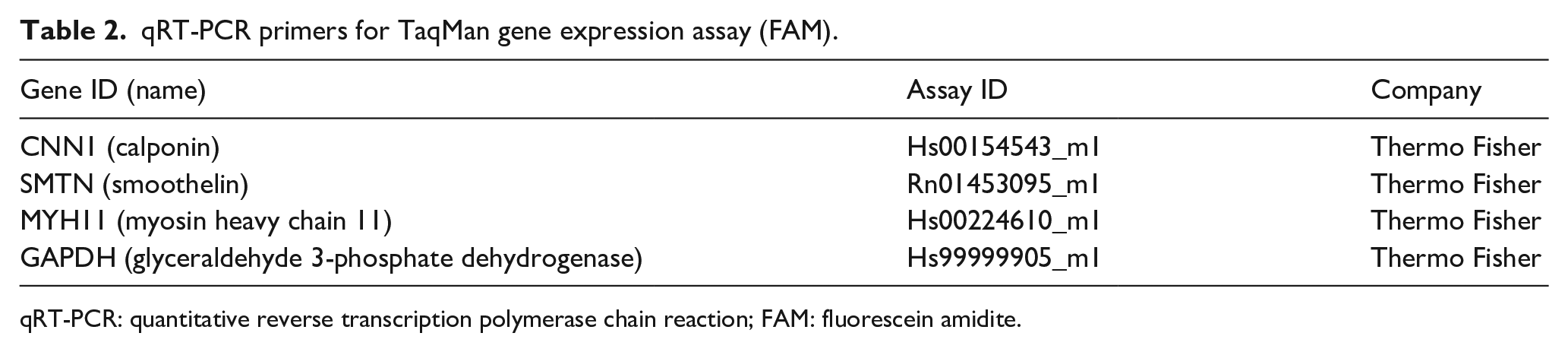
qRT-PCR: quantitative reverse transcription polymerase chain reaction; FAM: fluorescein amidite.
Statistics
Data are presented as mean ± standard error of the mean (SEM). Statistical analysis was performed using GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA) by one-way ANOVA and Tukey’s test. Statistical significance was defined as *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. Each data point represents a biological replicate, and all experiments were performed in technical triplicates.
Results
Cell characterization of SMCs, ADSCs, and pADSCs
SMCs demonstrated positive expression for α-SMA (96.33% ± 0.96), calponin (89% ± 1.8), smoothelin (80.1% ± 3.8), and MyH-11 (27.87% ± 4.41) (Figure 2(a)). Immunostaining with lineage-associated markers revealed that ADSCs were positively stained for CD29 (71.8% ± 5.7), CD44 (74.84% ± 12.1), CD73 (53.6% ± 3.6), and CD90 (95.5% ± 0.39 SEM) and negatively stained for CD34 (4.37% ± 1.5), and CD45 (6.33% ± 2.6) (Figure 2(b)). Under pre-differentiation conditions for 3 weeks, pADSCs exhibited expression of SMC-specific markers such as α-SMA (87.18% ± 2.45), calponin (81.02% ± 3.34), smoothelin (52.67% ± 1.4), and MyH-11 (17.45% ± 0.29), resembling bladder-derived SMCs (Figure 2(c)).
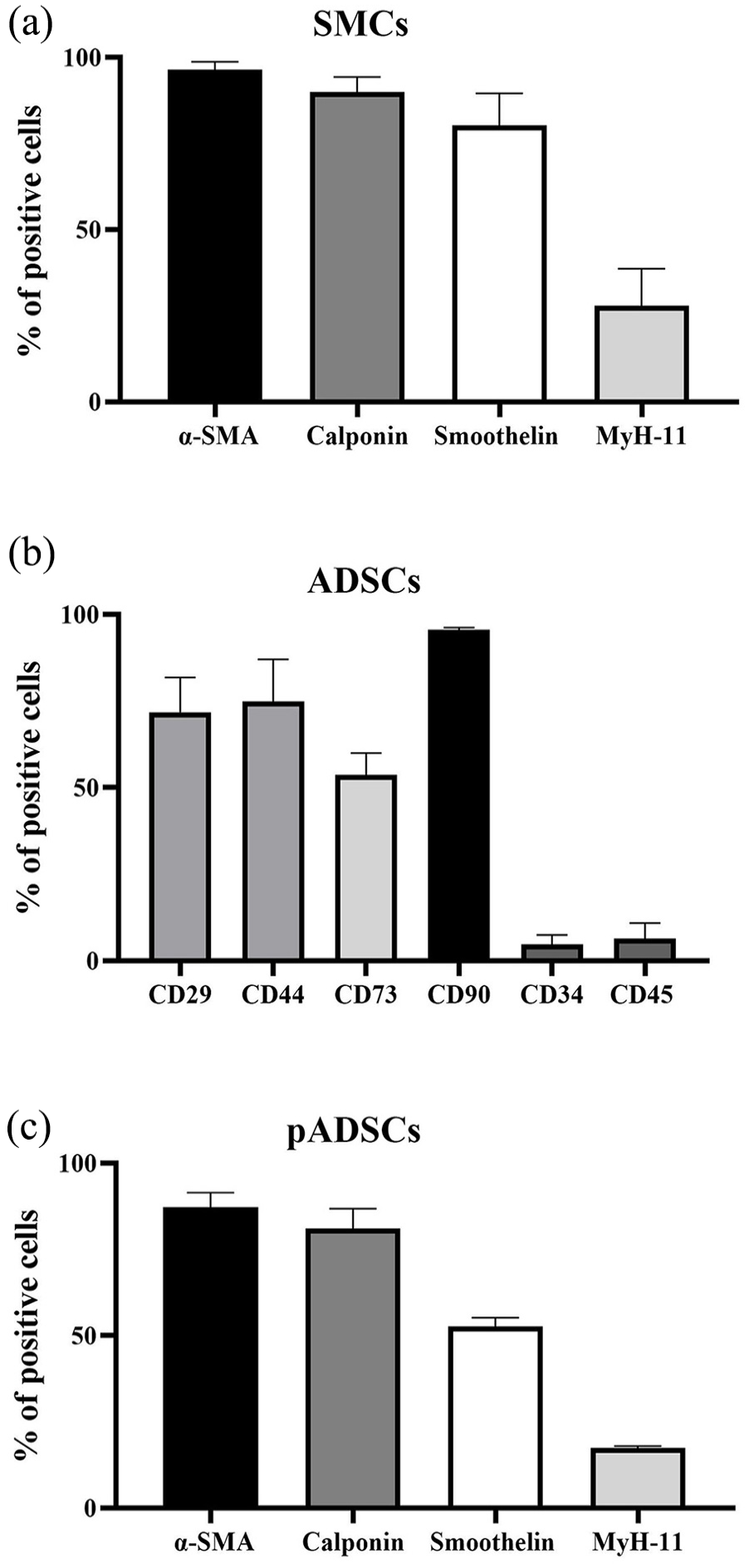
Characterization of rat ADSC, pre-differentiated ADSC, and bladder-derived SMCs. (a) SMCs (N = 6) were stained with α-SMA, calponin, smoothelin, and MyH-11. (b) Isolated ADSCs were stained for CD29, CD44, CD73, CD90, CD34 and CD45, and characterized by FACS (N = 3). (c) Pre-differentiated pADSCs (N = 3) were stained with α-SMA, calponin, smoothelin, and MyH-11.
CSCCS characteristics
All CSCCS proved to be mechanically stable tissue substitutes suitable for surgical handling such as suturing and stretching (Figure 3(a)–(d)).
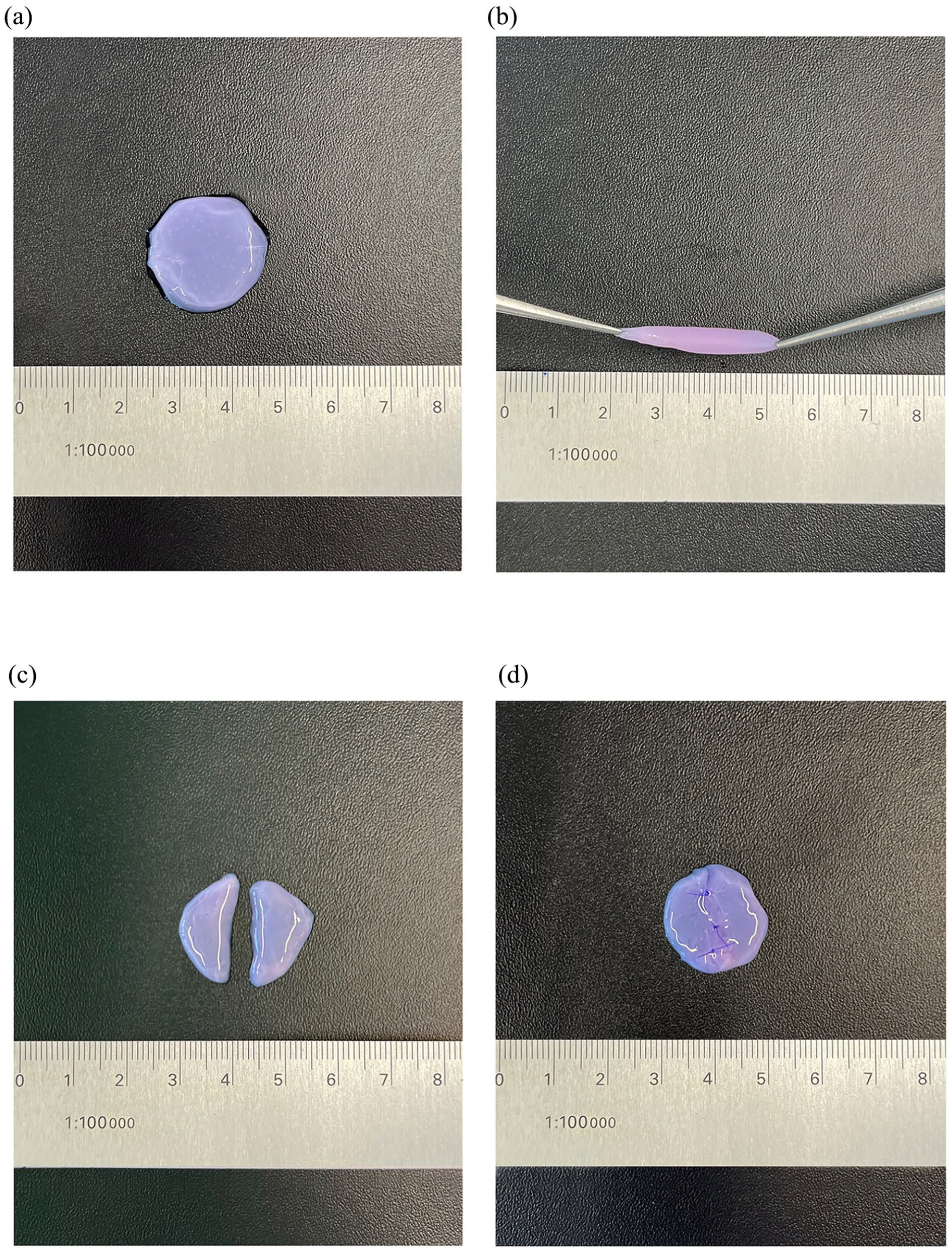
CSCCS characteristics. (a) CSCCS with scale comparison. (b) CSCCS can be stretched without tearing, representing crucial surgical handling properties, such as adjustment to wound margins. (c) A clean cut dividing the CSCCS was performed with surgical scissors. (d) The beforehand cut CSCCS was sutured back together using a surgical thread (6–0, braided).
After 2 weeks of incubation, the cell count within the camera FOV for the 2D SMC CSCCS was 6240 ± 442, for the 3D SMC CSCCS 9616 ± 249, for the 3D SMC/ADSC CSCCS 7602 ± 135, and for the 3D SMC/pADSC CSCCS 1512 ± 91. The 3D SMC CSCCS showed a significantly higher cell count than all other conditions (p < 0.001). The 3D SMC/pADSC CSCCS showed a significantly lower cell count than all other conditions (p < 0.001) (Figure 4(a)).
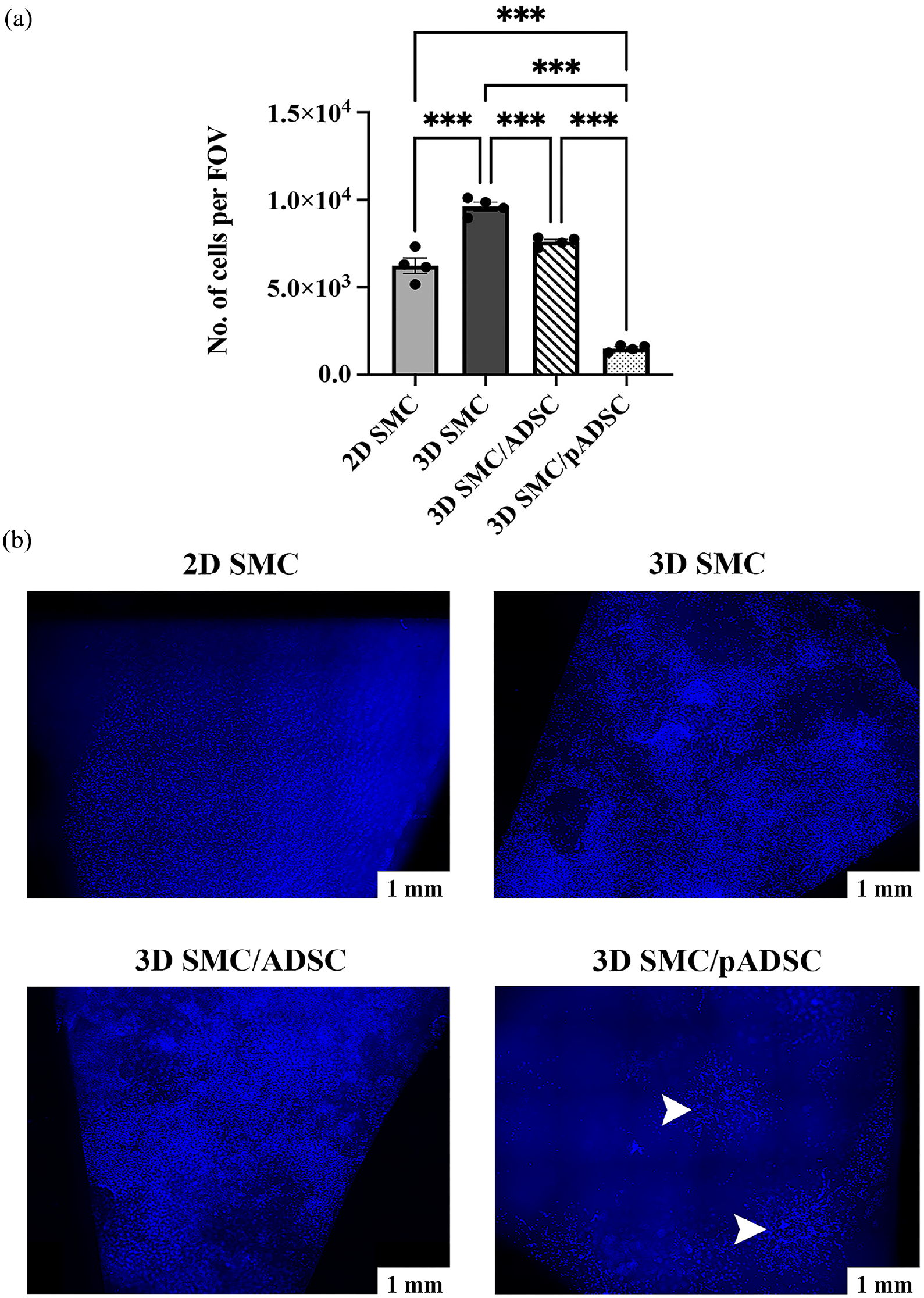
Cell count and cell distribution within CSCCS. (a) Cell count for each CSCCS was performed in triplicate in representative areas per 5× magnification camera field of view (FOV). 3D SMC monoculture showed a significantly higher cell count than all other conditions. 3D SMC/pADSC co-culture showed a significantly lower cell count than all other conditions. (b) DAPI-stained tile scans for comparison of the cell distribution within the CSCCS after 2 weeks of incubation. While 2D SMCs populate the CSCCS in an even pattern, 3D SMCs, and 3D SMCs/ADSCs populate the CSCCS in a patchy manner forming interconnected cellular networks. The three conditions show a sub-confluent scaffold population. Within the 3D SMC/pADSC CSCCS origins of spheroids were still detectable, and cells did not reach sub-confluency. Arrowheads point to spheroid origins. Scale bar is 1 mm.
Cell distribution was homogeneous in the 2D CSCCS. 2D SMCs spread as a thin two-dimensional layer. In contrast, the 3D SMCs grew out of the initially seeded spheroids in all directions and spread throughout the scaffold, forming a multilayered, interconnected cellular network. The 3D SMC/ADSC co-culture showed a similar distribution to the 3D SMC monoculture. However, within the 3D SMC/pADSC CSCCS, the 3D network was not as evenly distributed, and the cells tended to gather more around the former spheroids. Even after 2 weeks of incubation, most spheroids could still be identified as such within the network (Figure 4(b)).
SMC-specific contractile phenotype
SMC-specific contractile proteins calponin, smoothelin, and MyH-11 could be detected in all conditions.
In the 2D SMC CSCCS, the MFI for calponin, smoothelin, and MyH-11 measured after 2 weeks of incubation was 8188 ± 166, 2417 ± 241, and 1771 ± 45, respectively. In the 3D SMC CSCCS, the MFI for calponin, smoothelin, and MyH-11 was 6704 ± 734, 3990 ± 260, and MyH-11 2827 ± 264, respectively. In the 3D SMC/ADSC CSCCS, the MFI for calponin, smoothelin, and MyH-11 was 2474 ± 506, 1900 ± 230, and 774 ± 59, respectively. In the 3D SMC/pADSC CSCCS, the MFI for calponin, smoothelin, and MyH-11 was 1595 ± 526, 5912 ± 125, and 1531 ± 40, respectively (Figure 5(a)).
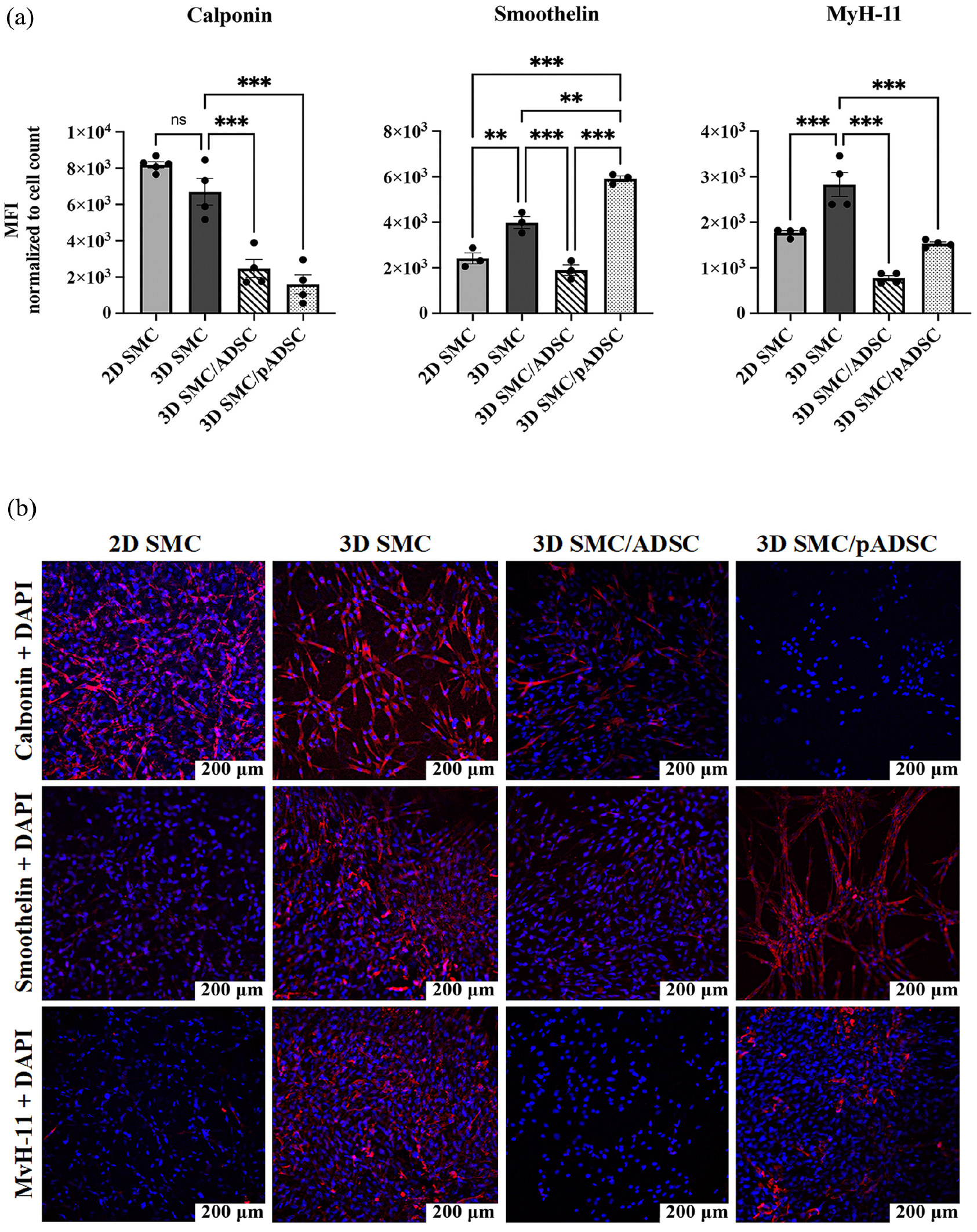
SMC-specific contractile phenotype. (a) Comparison of mean fluorescent intensity (MFI) of calponin, smoothelin, and MyH-11 divided by the associated cell count between the experimental conditions. 3D SMCs show a significantly higher expression of smoothelin and MyH-11 than 2D SMCs and 3D SMCs/ADSCs. 3D SMCs/pADSCs show a significantly higher expression of smoothelin than all other conditions. (b) Immunofluorescent Staining of the SMC-specific contractile protein markers calponin, smoothelin, and MyH11 as well as DAPI within the four scaffold conditions. The cells within the 2D SMC CSCCS show an intense fluorescent signal of calponin, followed by a steep decrease in the signal expression of smoothelin, and almost no expression of MyH11. In contrast, the cells within the 3D SMC CSCCS show an intense signal of MyH11 and smoothelin, followed by a slightly lower signal of Calponin. The cells of the 3D SMC/ADSC co-culture show a low signal of calponin and almost no signal of smoothelin and MyH-11. The cells derived from the 3D SMC/pADSC co-culture show an intense signal of smoothelin, followed by MyH11, and almost no fluorescent signal of calponin. Scale bar is 200 μm.
Calponin expression in the 3D SMC CSCCS was significantly higher compared to the 3D SMC/ADSC CSCCS (p < 0.001) and the 3D SMC/pADSC CSCCS (p < 0.001). Smoothelin expression in 3D SMC CSCCS was significantly higher compared to the 2D SMC CSCCS (p < 0.01) and the 3D SMC/ADSC CSCCS (p < 0.001). MyH-11 expression in the 3D SMC CSCCS was significantly higher compared to all other conditions (p < 0.001) (Figure 5(a)).
Smoothelin expression in the 3D SMC/pADSC CSCCS was significantly higher compared to the 2D SMC CSCCS (p < 0.001), the 3D SMC CSCCS (p < 0.01), and the 3D SMC/ADSC CSCCS (p < 0.001) (Figure 5(a)).
Fluorescent staining of the four experimental conditions is shown in Figure 5(b).
SMC-specific contractile gene expression
Examined SMC-specific contractile genes were expressed in every condition.
In the 2D SMC CSCCS, the gene fold relativized to GAPDH after 2 weeks of incubation for calponin, smoothelin, and MyH-11 was 0.046 ± 0.013, 0.0069 ± 0.00084, and 0.0012 ± 0.000048, respectively. In the 3D SMC CSCCS, the gene folds for calponin, smoothelin, and MyH-11 were 0.2 ± 0.017, 0.04 ± 0.027, and 0.036 ± 0.0013, respectively. In the 3D SMC/ADSC CSCCS, the gene folds for calponin, smoothelin, and MyH-11 were 0.042 ± 0.0021, 0.014 ± 0.0013, and 0.0025 ± 0.000056, respectively. In the 3D SMC/pADSC CSCCS, the gene folds for calponin, smoothelin, and MyH-11 were 0.16 ± 0.018, 0.029 ± 0.0011, and 0.026 ± 0.0021, respectively (Figure 6(a)).
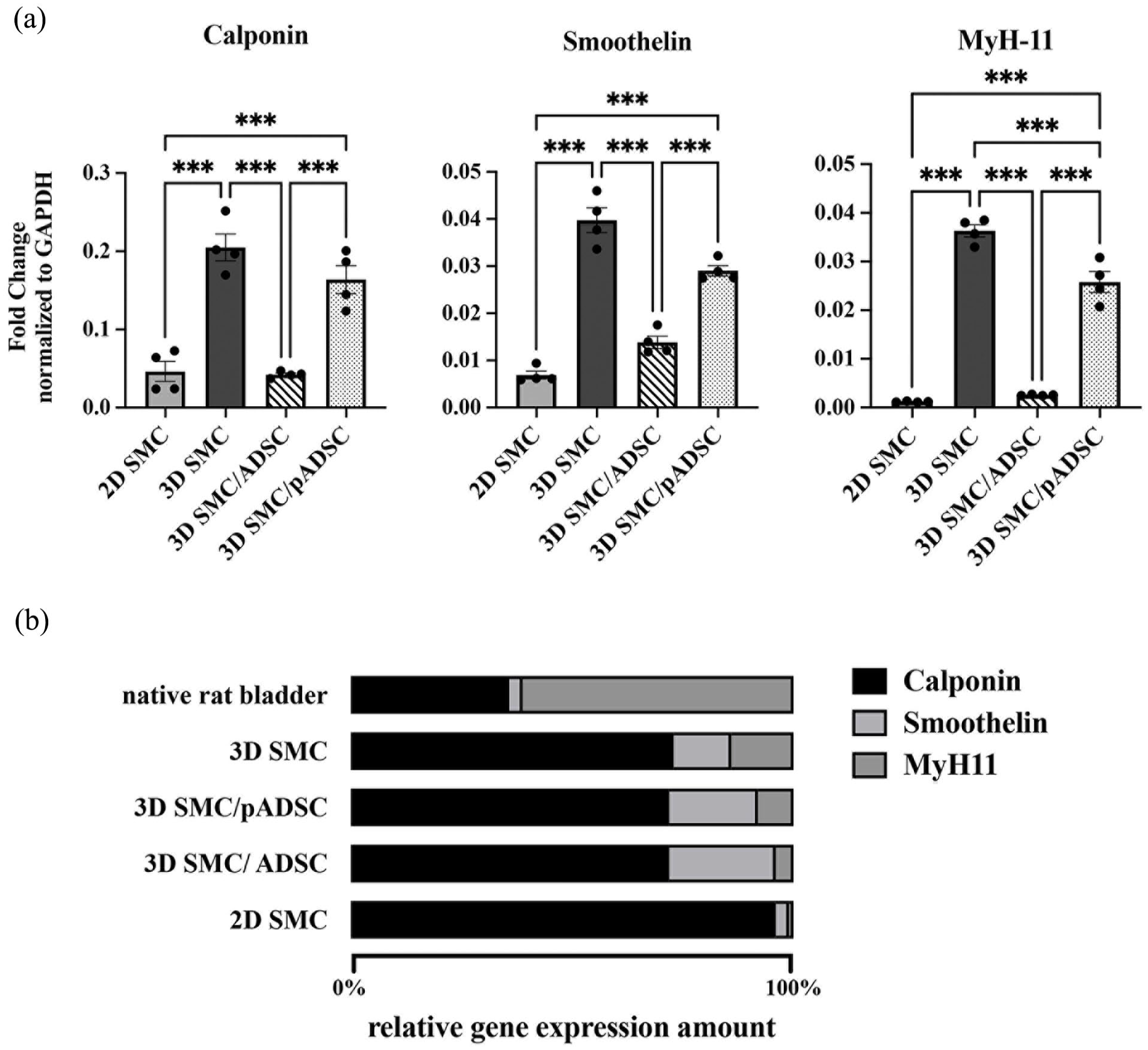
SMC-specific contractile gene expression. (a) Comparison of the fold change of the markers calponin, smoothelin, and MyH-11 normalized by each associated GAPDH value. The 3D SMC monoculture shows a distinctively higher gene expression of all three markers compared to all other conditions, followed by the 3D SMC/pADSC co-culture. (b) The relative gene expression amount within all four experimental conditions is shown in percent compared to each gene’s expression within the native rat bladder. The comparison shows how the composition of the 3D SMC CSCCS is closest to the composition of the native rat bladder.
Gene induction for calponin in the 3D SMC CSCCS was significantly higher compared to the 2D SMC CSCCS (p < 0.001), and the 3D SMC/ADSC CSCCS (p < 0.001). Gene induction for smoothelin in the 3D SMC CSCCS was significantly higher compared to the 2D SMC CSCCS (p < 0.001), and 3D SMC/ADSC CSCCS (p < 0.001). Gene induction for MyH-11 in the 3D SMC CSCCS was significantly higher compared to the 2D SMC CSCCS (p < 0.001), the 3D SMC/ADSC CSCCS (p < 0.001), and the 3D SMC/pADSC CSCCS (p < 0.001). Gene induction for calponin, smoothelin, and MyH-11 in the 3D SMC/pADSC CSCCS was significantly higher compared to the 2D SMC CSCCS (p < 0.001) and the 3D SMC/ADSC CSCCS (p < 0.001) (Figure 6(a)).
Native rat bladder tissue showed a gene expression pattern with the highest amount of MyH-11, followed by calponin and smoothelin. All CSCCS showed deviating patterns from the natural template. The CSCCS displayed an abundance of calponin expression, with a tendency towards higher MyH-11 expression and lower calponin expression gradually from 2D SMCs over 3D SMCs/ADSCs and 3D SMCs/pADSCs to 3D SMCs (Figure 6(b)).
Discussion
The primary aim of this study was to assess whether 3D SMCs in compressed collagen excel as building blocks for bladder TE compared to 2D SMCs to provide a superior cell-seeded collagen scaffold. While 3D SMCs already serve as in vitro disease models, seeding primary, bladder-derived SMC spheroids into a collagen scaffold to generate a pre-formed bladder wall for bladder TE purposes represents a novelty. By using an established compressed collagen scaffold and 3D SMCs we now have a tissue-engineered bladder scaffold with highly functional cells, which can be used for further in vivo bladder TE experiments. We also explored the potential of ADSCs and pADSCs as supporting cell sources and their impact on 3D SMC proliferation and differentiation. Our main hypothesis regarding the superiority of 3D SMCs in a collagen hydrogel over 2D SMCs in terms of contractile potential indicated by the expression of contractile genes and proteins was confirmed. Furthermore, the results of the 3D SMC distribution and proliferation within the CSCCS were favorable over 2D SMCs. However, 3D SMC/ADSC co-culture did not enhance SMC proliferation or contractile marker expression within the CSCCS. While 3D SMC/pADSC co-culture exhibited higher gene expression of all measured contractile markers as well as higher protein expression of smoothelin than the 2D SMC monoculture, it ultimately fell short of the 3D SMC monoculture.
The expression of contractile proteins serves as an indicator of cell differentiation and indirectly reflects cellular functionality. 39 While α-SMA is a non-specific marker expressed by SMCs, myofibroblasts, and fibroblasts, calponin is more specific, being associated with myogenic development. 39 Smoothelin and MyH-11 exhibit high specificity, with MyH-11 considered the most definitive marker of differentiated SMCs.39–41 Unfortunately, both smoothelin and MyH-11 protein expression diminish rapidly in 2D cultured SMCs, indicating de-differentiation and loss of function.16,42 A study by Huber and Badylak revealed that rat bladder SMCs from conventional 2D in vitro expansion stained positive for calponin but negative for smoothelin. 16 Babij et al. 43 observed a significant decrease in MyH-11 protein expression in cultured smooth muscle cells from rat aorta. In established human SMC lines, MyH-11 is either not expressed or shows low expression.44,45 In vivo, SMC phenotypes vary along a continuum between highly contractile, proliferative quiescent, and highly proliferative with low contractility, depending on their tissue-specific function. 39 This phenotype switching can occur in both directions, such as in response to injury.39,42,46,47 For bladder TE, the ideal scenario is highly proliferative SMCs initially populating the scaffold and subsequently transitioning to a quiescent, highly contractile phenotype. Chamley and Campbell demonstrated a phenotype switch in 2D culture, where densely seeded SMCs formed “hills and valleys” after reaching confluence, re-acquiring spontaneous contraction, indicating re-differentiation.42,48,49 However, a full recovery to the original in vivo phenotype was not observed. 50 In 2021, Gerwinn et al. 25 demonstrated that bladder SMC spheroids predominantly expressed the late myogenic differentiation marker MyH-11, unlike conventionally cultured 2D SMCs. A follow-up study confirmed that SMCs from diseased human bladder biopsies, when cultured as spheroids, regenerated their contractile phenotype and genotype. 26 This aligns with prior research indicating that spheroid cell cultures generally exhibit improved biological properties, such as increased proliferative activity and physiological metabolic function. 20 Notably, Jäger et al., 51 studying vascular SMC spheroids, observed downregulation of genes associated with proliferation, promoting cellular quiescence and indicating more stable differentiation markers than their 2D counterparts.
Our data affirm the superior attributes associated with spheroid cultures. Cells derived from 3D SMCs maintain an enhanced contractile phenotype over 2D SMCs when seeded in a compressed collagen scaffold, exhibiting increased presence, particularly of smoothelin and MyH-11, indicative of mature and contractile SMCs. Gene expression analysis demonstrated significantly higher expression of all examined contractile genes in 3D SMCs compared to 2D SMCs, highlighting their superior contractile potential—an essential characteristic for a functional bladder substitute. Furthermore, the gene expression trend observed in 3D SMCs suggests a restructuring toward the natural template, aligning with literature stating that spheroid cultures display a genotype closer to in vivo characteristics.52,53 This implies that 3D SMCs are more differentiated than their 2D counterparts and, according to their gene and protein expression profile, appear closer to a proliferative quiescent but highly contractile phenotype along the continuum of cell differentiation. In contrast, 2D SMCs seem closer to a highly proliferative but less contractile phenotype. We conclude that not only do 3D SMCs express more stable differentiation markers, as observed by Jäger et al., 51 but they also maintain their superior proliferative capacity—an attribute known for spheroid cultures. Importantly, the 3D CSCCS yielded a significantly higher cell count per camera field of view compared to the 2D SMCs, suggesting that 3D SMCs maintained proliferative capacity while exhibiting a stronger contractile genotype and phenotype than their 2D counterparts. This raises the possibility that SMC spheroids can undergo a phenotype transition. As they emerge from the spheroids to colonize the CSCCS, 3D SMCs appear to adopt a proliferative phenotype. However, upon attaining confluence within the scaffold, these 3D SMCs shift back to their contractile phenotype and intensify protein translation. 2D SMCs exhibit limited expression of contractile marker genes. After 2 weeks of incubation, SMCs derived from 3D culture had spread out from the spheroids, populating the entire scaffold with a three-dimensional network. In contrast, 2D SMCs organized themselves in a thin, two-dimensional layer. The distribution is crucial for generating a homogeneous, robust bladder. A sparse cell distribution within the scaffold reduces cell-cell signaling and minimizes scaffold compaction, while higher cell densities enhance extracellular matrix protein expression, reinforcing mechanical properties. 54 Moreover, complete scaffold colonization is essential, as post-implantation, nutrient supply to the tissue substitute occurs through diffusion from cell to cell in the capillary bed.
To address the challenge of obtaining sufficient cells from pathologically altered bladders, we explored the use of ADSCs and pADSCs. Including ADSCs did not significantly enhance overall regeneration compared to 3D SMC cultures. The 3D SMC/pADSC co-culture demonstrated elevated protein levels of smoothelin compared to the 3D SMC monoculture, along with increased gene expression of all measured markers compared to the 2D SMC monoculture. It is noteworthy that the proliferation within the 3D SMC/pADSC CSCCS was markedly lower than observed in the other conditions.
Several studies have demonstrated the positive impact of ADSCs on bladder SMC regeneration. Tremp et al. revealed that injecting ADSCs into the bladder wall improved voiding pressures and volumes in a rodent bladder outlet obstruction model, attributed to decreased fibrotic remodeling. 55 The upregulation of calponin, smoothelin, and MyH-11 on gene and protein levels within the smooth muscle bundles was linked to ADSCs paracrine effects aiding smooth muscle regeneration. 55 Zhu et al. 56 illustrated that ADSCs seeded onto bladder acellular matrix grafts for bladder reconstruction in a rabbit model promoted cell ingrowth and SMC regeneration. Our approach to co-culture SMCs and pADSCs on the spheroid level was inspired by our group’s recent study, demonstrating that the combination of SMCs and pADSCs led to increased proliferation and differentiation compared to each cell type alone. 31 We further observed that pADSCs play a crucial role in spheroid formation when co-cultured with SMCs in a conventional 2D fashion. 31 In our previous study, SMCs and pADSCs organized with SMCs forming the core, while pADSCs grew around this core, interpreted as a feeder layer or supporting cell layer. 31 The same development was observed with cells grown in collagen scaffolds, where pADSCs surrounded the core formed by SMCs. 31
The absence of an observed effect of ADSCs on SMC proliferation or contractile potential in our current study can be justified as follows. Spheroid assembly occurs partly through homophilic cadherin-induced tight cell connections.57,58 Co-culturing ADSCs with SMCs in spheroids possibly reduced SMC-SMC interaction, which may have reduced the benefits of SMC spheroid monoculture observed in our previous studies. 25 Jun et al. 59 found in a spheroidal co-culture model that individual cell function was strongly influenced by the ratios used, suggesting that adjusting the SMC-ADSC ratio during spheroid generation could yield different results. Stem cell spheroids generally promote regeneration by enhancing differentiation and paracrine effects post-transplantation. 57 ADSCs, in particular, are known to decrease bladder wall fibrosis, enhance cell migration, and promote angiogenesis through paracrine signaling in animal models,27,29,30,55,60 with benefits more evident in vivo. Whether this will also be the case in our application will have to be tested in vivo. In any case, the present in vitro model is suitable for investigating the SMC/ADSC interaction at the molecular level.
Regarding the use of pADSCs as an alternative cell source, we observed an improved contractile genotype in the 3D SMC/pADSC co-culture compared to the 2D SMC monoculture and the 3D SMCS/ADSC co-culture, along with the highest smoothelin expression. This is in line with our previous findings of decreased MyH-11 expression in pADSCs, likely due to the more immature smooth muscle phenotype after 3 weeks of differentiation. 31 However, pADSCs showed reduced proliferative potential and scaffold integration, maintaining spheroid formation and a significantly lower cell count per field of view. Earlier, our group observed SMC spheroid formation in direct co-culture with pADSCs, 31 where after 4 days, pADSCs showed higher proliferation rates than SMCs, with SMC proliferation only increasing in non-contact transwell co-culture. 31 Direct co-culture of SMCs and pADSCs in a collagen scaffold led to spheroid formation, where SMCs were surrounded by pADSCs, 31 likely impeding SMCs. As observed by Jäger et al., 51 cells in vascular SMC spheroids enter quiescence, suggesting that the outer cadherin-linked pADSC layer may reduce SMC proliferation and outgrowth from the spheroids into the scaffold. Huang et al., 61 who analyzed metastatic and non-tumorigenic breast cancer cells co-culture spheroid invasion within a collagen matrix, noticed an architecture-dependent regulation of tumor invasion, where metastatic cells completely enclosed by non-tumorigenic cells showed decreased cell invasion into the collagen matrix. 61 While maintenance of spheroid structures within the scaffold is beneficial for cancer research, 62 bladder TE requires dense, interconnected cell distribution. To harness pADSCs as an alternative cell source for bladder TE, it should be investigated whether it would be advantageous to cultivate spheroids separately before seeding them into the scaffold to avoid the effect described above.
Our study aimed to create a cell-seeded scaffold containing highly functional SMCs superior to 2D SMCs. Our group previously tested a cell-seeded compressed collagen hydrogel with promising outcomes in vivo. 12 Furthermore, we recently showed that 3D SMCs from rodent detrusor muscles express higher levels of contractile markers, show enhanced differentiation, and improved contractile potential compared to 2D SMCs. 25 The current study combined our established collagen scaffold with 3D SMCs and proved that SMCs cultured as spheroids maintain their superior qualities over conventionally cultured SMCs even after scaffold integration. These findings support advancing to in vivo trials with our CSCCS model.
Challenges arose in directly quantifying contractile protein levels through automated western blot analysis due to interference caused by the high collagen content in the CSCCS. Consequently, we had to resort to an indirect quantification method based on mean fluorescence intensity (MFI) normalized by cell count. It is crucial to highlight that the results obtained using this approach are representative of specific areas rather than the entire tissue substitute.
Conclusion
3D SMCs in compressed collagen are superior building blocks for bladder tissue engineering, marking a significant step toward developing bladder replacement tissue for future clinical applications. In comparison to conventional 2D cultures, they densely populate the scaffold, maintaining an enhanced phenotype and genotype. This suggests their potential for clinical translation in bladder tissue engineering. While ADSCs did not contribute further to improving cell proliferation and maturation of 3D SMCs in vitro, pADSCs might serve as an additional cell source, especially when SMCs from biopsies are limited. Overall, this study provides a cell-seeded scaffold with promising cell qualities and regeneration for further bladder TE experiments.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Baugarten Foundation (Zurich, Switzerland); Children’s Research Center (Zurich, Switzerland), and the Fromm Fellowship (Zurich, Switzerland).
Ethical statement
Experiments were conducted following the ethical guidelines for the use of animals in research (license no. 110/2015, Ethics Committee for Animal Experimentation, ECAE).