
Abstract
Numerous studies have addressed the use of vancomycin (VA) to effectively treat bacterial infections. However, VA is known to cause side effects when administered intravenously. Herein, monodisperse poly(N-isopropylacrylamide) (PNIPAAm) hollow nanocapsules were synthesized at the interface of a water-in-oil (W/O) single emulsion via Shirasu porous glass (SPG) membrane emulsification and UV-initiated polymerization. In water solutions, the PNIPAAm nanocapsules were able to encapsulate VA and form a new nanoscale water-soluble drug delivery system, namely, PNIPAAm-VA. In vitro experiments showed that PNIPAAm and PNIPAAm-VA had no cytotoxicity toward human bone marrow mesenchymal stem cells. In addition, the slow hydrolysis of PNIPAAm-VA in vitro led to the progressive release of VA, which was discharged at more than 50% and 80% of its initial concentration within 10 days at 37°C and 40°C, respectively; this subsequently inhibited the growth of methicillin-resistant Staphylococcus aureus bacteria. We believe that our PNIPAAm-VA nanoparticles can potentially be used as an effective injectable for temperature-sensitive materials in vivo to achieve the localized controlled release of drugs as safe and specific therapeutic agents.

Introduction
In recent years, the incidence of infection after surgical incision has been decreasing, but it still accounts for approximately 0.0218% of the total infections, and 44% of these cases involve Staphylococcus aureus infections. 1 The poor blood supply that accompanies this type of infection limits the delivery of antibiotics to the affected site, making infection prevention and control extremely difficult. 2 Moreover, after surgeries that involve medical implant insertion, microbial biofilms can easily form on the implant surface, further increasing the risk of infections that are difficult to treat, and it often causes a local tissue temperature increase of 1℃–2℃. 3 Commonly, these infections are treated using long-term standardized oral or intravenous antibiotics. 4 For most patients, despite systematic and accurate treatment, it is still possible to develop severe infection due to poor blood flow to the target site, which does not allow a sufficient concentration of antibiotics to reach the site of interest. In addition, repeated exposure to high doses of antibiotics can lead to increased bacterial resistance and consequently result in systemic toxicity. 5 Therefore, localized drug delivery systems are being developed to not only reduce side effects but also enhance the treatment efficacy by releasing antibiotics near the infected site. 6 At present, several drug carriers for localized drug delivery, such as naturally occurring minerals (calcium sulfate, hydroxyapatite) or synthetic polymers (such as polymethylmethacrylate (PMMA) and poly(lactic-co-glycolic acid) (PLGA)7 –11 have been developed. Notably, some polymers have exhibited the capacity to locally deliver drugs at high concentrations with low systemic distributions. However, the main disadvantage of these systems is that they require invasive implantation to be inserted into the infected site, which is often accompanied by additional medical risks and complications, such as pain and the chance of infection as well as high costs. Ideally, localized drug delivery to the target site is applied without invasive implantation. 12
The temperature-responsive behavior exhibited by poly (N-isopropylacrylamide) (PNIPAAm) in water solutions has garnered great interest in research.13 –15 PNIPAAm undergoes a reversible phase transition at a lower critical solution temperature (LCST) of approximately 32°C. When the temperature is below the LCST, PNIPAAm is hydrophilic, with swollen and hydrated polymer chains. When the temperature is above the LCST, the hydrogen bonds between water molecules and the hydrophilic moieties of the polymer are disrupted, thereby achieving thermosensitive drug delivery. 16 In addition, the LCST of PNIPAAm can be modified by the addition of surfactants, salts and other polymers to the solutions or by chemical modification using a variety of agents. In particular, copolymerization with hydrophilic molecules can increase the LCST, while copolymerization with hydrophobic monomers can decrease the LCST. 17 This makes PNIPAAm one of the most temperature-sensitive materials.18,19
Vancomycin (VA) is a glycopeptide antibiotic that functions by inhibiting bacterial cell wall synthesis, thus significantly inhibiting the growth and biofilm formation of gram-positive bacteria. 20 Currently, VA is one of the most commonly used drugs in bacterial infection prevention and treatment and has been found to be effective against methicillin-resistant Staphylococcus aureus bacterium.21 –24 However, it can also cause some side effects, such as nephrotoxicity and ototoxicity, after intravenous injection at high doses and may lead to bacterial resistance (e.g., the development of heterogeneous vancomycin-intermediate S. aureus).25,26 In particular, for surgical wounds with low accessibility (such as spinal surgery wounds), an option is to apply VA locally instead of injecting it. This method has proven to be effective in reducing the rate of infection after surgical incision via the use of a VA-based powder. 27
In light of the promising benefits of localized drug delivery, we synthesized VA-containing PNIPAAm nanoparticles (PNIPAAm-VA), which could potentially form in the body at room temperature after solution injection in a temperature-responsive manner, for the optimal controlled release of VA. Moreover, our technology could allow bacterial resistance to be overcome using an intelligent antimicrobial drug delivery system. In this study, nanoparticles were synthesized with the sol-gel process, and their morphology, structure, and physical/chemical properties were thoroughly analyzed. We determined the VA content in the nanoparticles, the VA release pattern in phosphate buffered solution at 37°C (PBS, pH = 7.4), and the biocompatibility and specificity of the delivery system against Staphylococcus aureus
Materials and methods
Materials
PNIPAAm was purchased from Soochow University (Suzhou, China). Dialysis bags (MWCO 2000) were purchased from Viskase (Lombard, IL, USA). Human bone marrow mesenchymal stem cells (BMSCs) were purchased from Bluefbio (Shanghai, China). VA was supplied by VIANEX S.A. (produced in plant C, Pallini, Greece). Methicillin-resistant Staphylococcus aureus (MRSA) was provided by The First Affiliated Hospital of Harbin Medical University (Nangang, China). Mueller-Hinton (MH) broth was purchased from Qingdao HopeBio-Technology Co., Ltd. (Quindao, China). Fetal bovine serum was purchased in Shanghai. DMEM was purchased from Sigma Co., Ltd. PBS solution was purchased from Qingdao Hope Biotechnology Company Ltd.
Synthesis of PNIPAAm and PNIPAAm-VA
Monodisperse PNIPAAm hollow nanocapsules were prepared at the interface of water-in-oil (W/O) single emulsions via Shirasu porous glass (SPG) membrane emulsification and UV-initiated polymerization. 28 First, in a typical emulsification process, 0.154 g N,N’-methylenebisacrylamide (MBA) crosslinker was added to a 20 ml aqueous solution of 2.26 g N-isopropylacrylamide (NIPAAm) monomer to form the disperse phase (water phase). A 320 ml kerosene solution containing 5 wt% polyglycerol polyricinoleate (PGPR) surfactant was used as the continuous phase (oil phase). Under the action of active free radicals produced by the decomposition of 2-dimethoxy-2-phenylacetophenone (BDK) in the continuous phase, a monodisperse monomer-containing W/O emulsion was prepared by emulsion molding of the SPG membrane. 29 Then, the NIPAAm monomer and crosslinker were encapsulated in monodisperse W/O single emulsions, and then the dispersed phase was injected through a porous membrane, with the resulting droplets forming at the openings of pores on the membrane surface after coming into contact with the continuous phase. Emulsification was carried out at room temperature (23°C). 30
UV polymerization was carried out at 20°C for 40 min in a nitrogen atmosphere. During UV irradiation, the prepared W/O emulsions and 20 ml kerosene containing 40 mg BDK were poured into a vessel, and then the mixture was gently stirred with magnetic rods to prevent particle coagulation. The polymerized nanocapsules were separated from the oil phase by 2000 rpm centrifugation for 10 min and then centrifuged with an aqueous solution containing 5 wt% surfactant PGPR and pure water five times (2500 rpm, 20 min, and 23°C). Finally, the nanocapsules were redispersed in deionized water at room temperature for characterization or for the following reaction steps (Figure 1).

Molecular structures of VA and PNIPAAm.
Synthesis of PNIPAAm-VA
Due to the hollow microcapsule structure of PNIPAAm, VA could be encapsulated into microcapsules through electrostatic adsorption. To prepare PNIPAAm-VA, 5 mg VA was added into 2 ml of a 1 mg/ml aqueous solution of PNIPAAm nanoparticles. The solution was stirred at room temperature for 2 h; then, the mixture was added into a dialysis bag (MWCO = 2000). The bag was immersed in deionized water (50 ml) at 23°C and placed on a magnetic stirrer for 24 h. The deionized water was refreshed every 4 h. The PNIPAAm-VA solution was the residual solution.
Determination of VA drug-loading capacity and encapsulation efficiency
First, we recorded the UV‒Vis absorption of VA at 281 nm to construct a calibration curve. We verified that its linear range of detection was 0.5–10 mg/ml. We mixed 2 mg of PNIPAAm solution with 2 ml of deionized water, and then 5 mg of VA was added to prepare the PNIPAAm-VA nanoparticles. After stirring the mixture for 2 h, the obtained solution was transferred into a dialysis bag and added into 50 ml deionized water to undergo dialysis for 24 h (NWCO = 2000). The deionized water was refreshed every 4 h. Then, a UV/Vis spectrophotometer was used to determine the VA concentration in the solution in the dialysis bag at a wavelength of 281 nm. The drug-loading capacity (DLC) and drug-loading efficiency (DLE) were calculated using equations (1) and (2). At the same time, an aqueous solution of PNIPAAm nanoparticles without VA was used as the blank control to prevent the interference of PNIPAAm in the UV‒Vis measurements.


*All weights are reported in mg.
In vitro release of PNIPAAm-VA
To conduct PNIPAAm-VA drug release tests, we recorded three VA concentration gradients in PBS (pH = 7.4) at 23°C, 37°C, and 40°C. For each measurement, three PNIPAAm-VA solution groups were used (PV1 = 1 mg/ml PNIPAAm with 1 mg/ml VA, PV2 = 1 mg/ml PNIPAAm with 5 mg/ml VA, and PV3 = 1 mg/ml PNIPAAm with 10 mg/ml VA). Then, 5 ml of the abovementioned solutions were prepared. After stirring each mixture for 2 h, the obtained solution was placed into a dialysis bag. The dialysis bags were then immersed in 10 ml PBS buffer and stirred with a magnetic stirrer (100 rpm) at different temperatures (23°C, 37°C, or 40°C). At predetermined intervals (1 day), 10 μl of the liquid in the dialysis bag was extracted, and the same volume of fresh PBS buffer was added outside the dialysis bag every 4 h. Finally, the VA release amount was analyzed using an ultraviolet spectrophotometer at 281 nm. The VA release experiment was repeated three times, and the results were calculated using the average values and standard deviations. The change in the amount of VA released over time was then plotted.
Cell culture
Normal BMSCs were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% calf serum and 0.1% antibiotics (50 units/ml penicillin and 50 units/ml streptomycin) at 37°C under 5% CO2.
Biocompatibility of nanoparticles
Cell Counting Kit-8 (CCK-8) was used to determine the cytocompatibility of PNIPAAm and PNIPAAm-VA with BMSCs. 31 First, 200 μl of BMSCs (5000 cells/well) were seeded in a 96-well plate and incubated for 24 h in an incubator with 5% CO2 at 37°C. Then, multiple 200 μl PNIPAAm, VA, and PNIPAAm-VA solutions with different concentrations were prepared to replace the culture medium: PNIPAAm with a series of concentrations, VA with a series of concentrations (0.1, 0.25, 0.5, 0.75, 1, or 1.5 mg/ml), and PNIPAAm-VA with a series of concentrations (when the concentration of PNIPAAm was 1 mg/ml, the concentration of VA was 0.1, 0.25, 0.5, 0.75, 1, or 1.5 mg/ml). A (insert concentration, insert solvent) solution of PNIPAAm without cells was used as a blank control, and pure DMEM without cells was used as the negative control. After culturing for 24, 48, or 72 h, 200 μl of DMEM was used to dilute the nanoparticle solutions, and 10 μl of CCK-8 solution was added to each well. After another 4 h of incubation, the absorbance spectrum of each solution was measured using UV/Vis spectrophotometry at 450 nm. The measurement was repeated three times for each group.
In vitro antibacterial activity of nanoparticles
The antibacterial properties of PNIPAAm and PNIPAAm-VA nanoparticles against bacteria were determined using the microbroth dilution method. 32 First, 150 μl of the above prepared solutions of PNIPAAm and VA (1, 2, 4, 6, 8, or 10 μg/ml) or PNIPAAm-VA (with 1 μg/ml PNIPAAm and 1, 2, 4, 6, 8, or 10 μg/ml VA) was added to a 96-well plate. Then, 50 μl of a MRSA bacteria solution in MBH (approximately 1.0 × 107 CFU/ml) was added to each well. The plate was then incubated at 23°C, 37°C, or 40°C for 24 h. Eventually, 50 μl of bacterial suspension from each well was diluted (1000 times) and inoculated on an agar plate, which was incubated at 37°C for 24 h. The residual bacterial colonies were then counted to evaluate the decrease in the number of live bacteria. The viable count was determined and reported as log10 (CFU/ml).
To determine the long-term effectiveness of our drug delivery system, we added 200 μl of a PNIPAAm-VA solution (1 μg/ml PNIPAAm and 6 μg/ml VA) into a 96-well plate with 100 μl of a MRSA bacteria solution (approximately 1.0 × 107 CFU/ml). The plate was then placed in an incubator at 37°C for different time periods, ranging from 1 to 10 days. Periodically, 50 μl of solution was removed, diluted, and inoculated on a suitable agar plate, which was then incubated at 37°C for 24 h. The viable count was then determined and reported as log10 (CFU/ml).
Statistical analysis
SPSS 21.0 software was used to conduct a statistical analysis of the results. All quantitative data were presented as the means ± standard deviations (expressed as the relative standard deviations (RSDs)). The t test was used to analyze the significant difference between groups and experiment durations. A value of p < 0.05 was considered to be statistically significant.
Results and discussion
Morphology and structural characterization of PNIPAAm and PNIPAAm-VA
During radical polymerization, BDK decomposes into a continuous phase to produce a large number of active free radicals. These radicals diffuse through the W/O interface and penetrate the water phase of the W/O emulsion. Moreover, under UV light, the NIPAAm monomers that dissolve in the water phase can also diffuse in the W/O interface, where they are polymerized by the action of the free radicals. As a result, a cross-linked PNIPAAm layer forms at the interface of a droplet due to the action of the crosslinking agent. As the PNIPAM layer at the interface becomes thicker, the amount of free radicals that are able to enter the aqueous phase gradually decreases, and so does the number of free radicals that can diffuse back to the oil phase; this results in a gradual slowing of the polymerization rate.33,34 Eventually, when the thickness of the PNIPAAm shell is so large that the free radicals cannot diffuse at all, polymerization stops, resulting in hollow PNIPAAm nanoparticles. In our study, at 20°C, we observed an average shell thickness of approximately 3.3 μm. The TEM micrograph of a PNIPAAm microcapsule is shown in Figure 2(a). The hollow structure of the PNIPAAm nanoparticle was easily to see, displaying an almost uniform shell thickness in all directions.
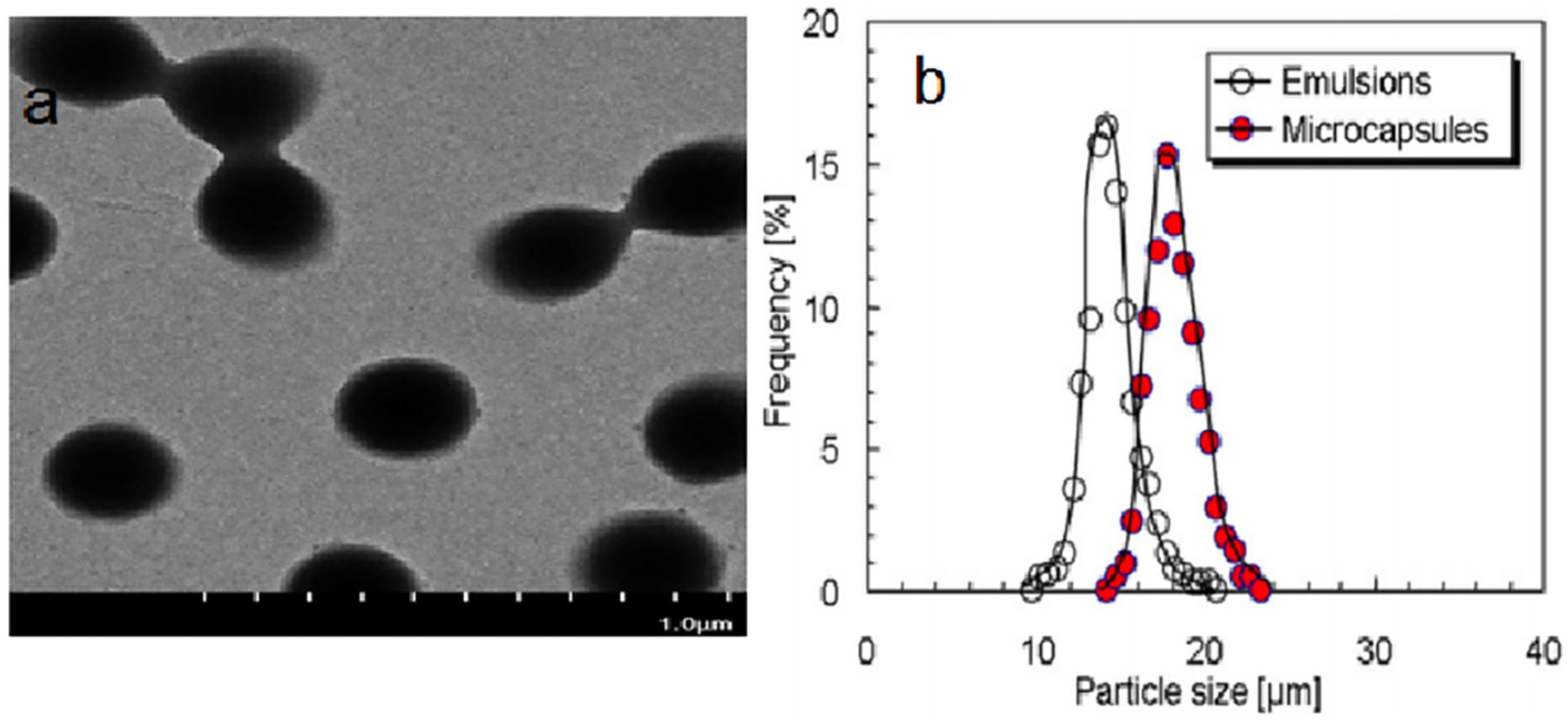
(a) TEM micrograph of a PNIPAAm-VA microcapsule, revealing a particle size of approximately 3.3 μm; (b) size distribution of W/O emulsion droplets and PNIPAAM microcapsules dispersed in water at 20°C.
Because the polymerization was carried out at a relatively low temperature (20°C) and the reaction was controlled, we were able to maintain the particle size dispersion coefficient (δ) before and after polymerization at approximately 0.27 (RSD 0.12, Figure 2(b)). The average particle size of the PNIPAAm nanoparticles was slightly larger than that of the W/O emulsion droplets containing the monomers. This was because the washed PNIPAAm nanoparticles showed a high expansion state in water at 20°C, which may have been related to the decrease in the half-life of the intramolecular hydrophobic interactions when the ambient temperature was lower than the LCST (approximately 32°C). 35
When VA was encapsulated in these PNIPAAm nanocapsules at 23°C, their size decreased from approximately 1.41 μm to 358 nm. During encapsulation, the electrostatic repulsion between the positively charged PNIPAAm molecules that constituted the nanocapsules decreased due to the incorporation of negatively charged VA molecules. As a result, the antibiotics and PNIPAAm molecules attracted each other, leading to a reduction in the size of the nanoparticle. In the normal in vivo environment, the permeability size of the blood vessel wall is approximately 200 nm, which means that after the local injection of PNIPAAm-VA nanoparticles, the particles still cannot travel in the body through the blood vessel wall, thus maintaining a local drug concentration.
VA loading capacity and encapsulation efficiency of PNIPAAm-VA nanoparticles
Initially, we recorded the absorbance spectra of VA and PNIPAAm nanoparticles at 281 nm (y = 0.449x − 0.0015, R2 = 1), and we constructed a calibration curve for VA. PNIPAAm nanoparticles without VA exhibited no obvious absorption at 281 nm. To determine the VA concentration in the nanoparticles, we measured the UV‒Vis absorbance of a PNIPAAm-VA solution after dialysis. Additionally, we substituted the obtained absorbance into the standard curve equation to obtain the corresponding VA concentration. Each experiment was repeated three times; according to the equation, the drug-loading rate and encapsulation rate of the nanoparticles were 53.6 (±0.3)% and 10.72 (±0.3)%, respectively. These results confirmed that the PNIPAAm nanoparticles have a high VA loading capacity and can encapsulate VA to be used as a modified drug-release carrier.
In vitro VA release of PNIPAAm-VA nanoparticles
According to changes in temperature and time, PNIPAAM-VA nanoparticles gradually release antibiotics. When the temperature exceeds the LCST of the nanoparticles, the microcapsules contract gradually, and the VA encapsulated in the particles is released gradually through electrostatic adsorption. When the temperature exceeds 40°C, the nanoparticles completely crack. In this work, we studied the antibiotic release kinetics of our PNIPAAm-VA system and observed that VA was continuously released for more than 10 days. We also noticed that the release kinetics were significantly temperature dependent. At higher temperatures, more VA was released: in 10 days, PNIPAAm-VA released approximately 30% VA at 23°C, 60% at 37°C, and 86% at 40°C (p ⩽ 0.05) (Figure 3). Moreover, a sudden release of VA was observed within the first 1–3 days. This may be due to the sudden contraction of the microcapsules and their supersaturated state leading to the release of a large amount of VA. Furthermore, the release of VA was related to its initial load. For higher initial loads of VA, the release rate was also higher. As shown in Figure 3(b), at 37°C, approximately 51% of VA was released from PV1 nanoparticles in 10 days, while PV2 and PV3 released 61% and 67% VA, respectively. We can conclude that encapsulating antibiotics in a hydrogel network can efficiently prolong the drug release period.
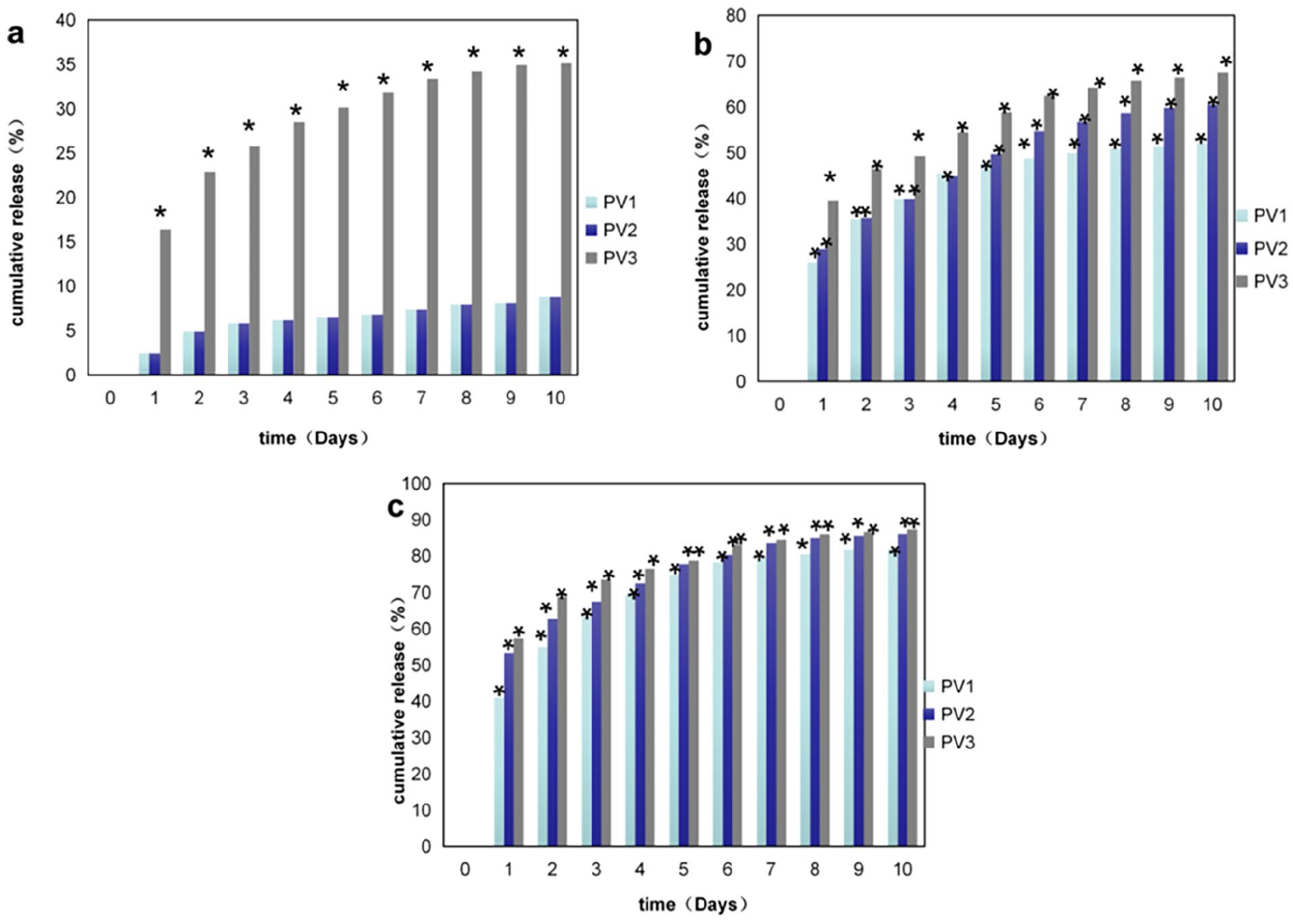
VA release rate of PNIPAAm-VA nanoparticles (PV1, PV2, and PV3) at 23°C (a), 37°C (b), and 40°C (c) test (p ⩽ 0.05). (PV1 = 1 mg/ml PNIPAAm with 1 mg/ml VA, PV2 = 1 mg/ml PNIPAAm with 5 mg/ml VA, and PV3 = 1 mg/ml PNIPAAm with 10 mg/ml VA).*P<0.05.
Biocompatibility
When developing new polymer-assisted drug delivery technologies, it is important to evaluate the potential toxicity of the carrier matrix. The CCK-8 assay was used to determine the cytotoxicity of PNIPAAm and PNIPAAm nanoparticles toward BMSC cells. This method is based on the ability of the mitochondrial dehydrogenases, present only in living cells, to reduce a provided salt to an orange formazan product. The number of living cells is in direct proportion with the amount of formazan produced, whose absorbance can be detected by UV‒vis spectro-photometry.
BMSC cells were cultured for 24, 48, or 72 h after adding different concentrations of PNIPAAm nanoparticles, PNIPAAm-VA nanoparticles, or VA. As shown in Figure 4(a), PNIPAAm nanoparticles showed good biocompatibility. Although cytoactivity decreased with increasing culture time, the cell survival rate remained above 80% after 72 h of culture. Similarly, we observed that both VA (Figure 4(b)) and PNIPAAm-VA nanoparticles (Figure 4(c)) showed good cytoactivity after being cultured for 24, 48, or 72 h (p < 0.05). These results suggested that the components of our system are not toxic to mammalian cells 36 because cell viability remained greater than 75% in every experiment. Based on these results, we can conclude that PNIPAAm-VA nanoparticles constitute a nontoxic and safe drug delivery system.

Cytoactivity of BMSCs after the addition of (a) PNIPAAm nanoparticles, (b) VA, and (c) PNIPAAm-VA at different concentrations for 24, 48, and 72 h. The values reported represent the means of three measurements (RSD = 6). No statistical difference between the groups (p = 0.05).
Antibacterial properties of PNIPAAm-VA nanoparticles
The antibacterial properties of our nanoparticles were evaluated by incubating a bacterial suspension with different PNIPAAm-VA solutions and evaluating whether the contact between the nanoparticles and bacteria resulted in a bactericidal effect. The minimum inhibitory concentration (MIC) of VA is 1–2 μg/ml. 37 According to the microbroth method, 150 μl of PNIPAAm nanoparticles, PNIPAAm-VA nanoparticles, or VA solutions (at different concentrations) was added into a 96-well plate; after diluting with 50 μl of MRSA solution (approximately 1.0 × 107 cfu/ml), they were cultured at 37°C for 24 h and then inoculated on an agar plate. The experimental results showed that PNIPAAm nanoparticles did not exhibit bacteriostatic properties against MRSA bacteria (Figure 5(a)). Interestingly, at 37°C, VA completely inhibited the growth of MRSA bacteria at 6 mg/ml (Figure 5(b)), while the VA-loaded nanoparticles only achieved bacteriostatic effects when the VA concentration reached 8 mg/ml (Figure 5(c)). This result showed that after the same addition of VA, when only the VA solution was added to achieve the bacteriostatic effect, the PNIPAAm-VA solution required an increase VA concentration to achieve a complete bacteriostatic effect. This result further confirmed that PNIPAAm can encapsulate VA and release it slowly.

Antibacterial activities of PNIPAAm, VA, and our nanoparticles. (a)–(c) MRSA bacteria (initial concentration = 1.0 × 105 CFU/ml) cultured for 24 h in solutions of different concentrations of PNIPAAm, VA and PNIPAAm-VA (p > 0.05).
We were also interested in evaluating the VA release rate at different temperatures; the results are shown in Figure 6(a). As the temperature increased, the amount of released VA gradually increased. For example, at 23°C, the PNIPAAm-VA nanoparticles only started to show antibacterial properties when the VA concentration was 10 μg/ml, while at 40°C, the bacterial viability was already exhausted. Moreover, we investigated the long-term antibacterial effect of the PNIPAAm-VA nanoparticles. A MRSA bacteria solution was added to a solution of PNIPAAm-VA nanoparticles, and the resulting mixture was cultured in a 37°C incubator for 1–10 days and then inoculated in an agar plate to determine the number of viable bacteria. The results are shown in Figure 6(b). As the time increased, the number of viable bacteria decreased, which confirmed that our PNIPAAm-VA nanoparticles can continuously release VA and inhibit the growth of MRSA over time. In conclusion, we proved that the VA loading and release of the PNIPAAm-VA nanoparticles constitutes a smart and effective antibacterial system with time-controlled drug release.

VA release rate and at different temperatures. (a) bacterial viability after culturing with PNIPAAm-VA nanoparticles at 23, 37 and 40°C for 24 h (p > 0.05); (b) drug release of PNIPAAm-VA nanoparticles over 10 days. All the data are reported as the mean values of three measurements (RSD = 6) (p > 0.05).
Conclusions
As the rate of bacterial drug resistance increases, the rate of MRSA infection also gradually increases. While it is effective to intravenously administer large doses of antibiotics, some adverse side effects inevitably occur, such as liver and kidney function damage. At the same time, as the local blood supply is insufficient during infection, it is difficult to achieve a high concentration of antibiotics in the infected site, which further increases bacterial drug resistance. In this paper, we investigated a prototype of a localized antibiotic injection method, which involved the controlled release of VA from PNIPAAm nanoparticles. A series of experiments established that this system has good biocompatibility, an extended drug release time, and an excellent sterilization effect. Moreover, we observed good temperature controllability because the amount of drug release increased with increasing temperature. This technology has a promising future as a noninvasive anti-infection system and can be applied in the treatment of infection in vascular tissues. However, this study only measured the relevant experimental features of PNIPAAm-VA nanoparticles in vitro, and further in vivo experiments have yet to be performed.
Footnotes
Acknowledgements
I would like to thank Chen Rui of Suzhou University Industrial Park and Harbin Veterinary Research Institute for their help.
Authors’ contributions
JS and LL designed all the study. SLL and XFK performed the data processing and experimental analysis. LL drafted the manuscript. All authors reviewed and approved the final version of the manuscript. LL, XFK, and SLL contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Heilongjiang Provincial Health Commission scientific research topic (2020-250).