
Abstract
Mitragyna speciosa (Korth.), commonly known as kratom, contains a variety of secondary metabolites, with mitragynine, an indole alkaloid, being the most significant due to its analgesic properties. However, the limited commercial availability of mitragynine standards highlights the need for effective separation techniques. The complexity of kratom’s plant matrix makes selective isolation of mitragynine challenging. This study investigates the use of dummy template molecularly imprinted polymers (MIPs) as selective sorbents for isolating mitragynine. Colchicine was selected as a promising dummy template molecule for mitragynine. Prepolymerization studies, including association constant and Job plot analysis, identified methacrylic acid in methanol as the optimal component for synthesis. MIP synthesis was optimized using the bulk polymerization method with a template-monomer-crosslinker ratio of 1:3:20, at 80°C for 24 hours. Template removal was achieved through sonication, employing a 16-cycle methanol, and infrared spectroscopy confirmed successful template removal. MIP adsorption followed a pseudo-second-order kinetic model and a Langmuir isotherm. Kratom extraction was performed using the reflux method with methanol. Separation of mitragynine was conducted via column chromatography with MIP as the sorbent, resulting in enhanced mitragynine isolation. Quantitative analysis showed that the MIP method achieved an isolation efficiency of 36.7%, whereas preparative thin-layer chromatography (PTLC) yielded only 3.607%. Meanwhile, visual observations using Thin-Layer Chromatography (TLC) demonstrated that MIP produced a cleaner chromatogram compared to conventional PTLC. These findings suggest that MIPs offer a promising alternative to conventional separation techniques for isolating mitragynine from complex plant extracts.
Keywords
Introduction
The kratom plant (Mitragyna speciosa) is extensively found in Southeast Asia, particularly in Indonesia. Kratom is frequently utilized in traditional medicine to address a range of conditions, including diarrhea, cough, muscular discomfort, and fatigue. 1 Kratom leaves possess several secondary metabolites, with mitragynine, an alkaloid, as the predominant constituent. 2 Kratom exhibits stimulating effects at low dosages and sedative effects at elevated doses, with mitragynine and 7-hydroxymitragynine, secondary metabolites produced by kratom, possessing anti-inflammatory, antidepressant, analgesic, psychotropic, and opioid properties. 3 As a result, the psychoactive and addictive properties of this plant have prompted the National Narcotics Agency (BNN) Indonesia to classify mitragynine as a new psychoactive substance (NPS), advocating for its designation as a Class I narcotic. Narcotics that are categorized as Class I are not permitted for medical use due to their potential for dependency and are exclusively appropriate for scientific research purposes. Despite its pharmacological importance, obtaining a standard reference of mitragynine remains a significant challenge due to the complexity of isolating this compound from kratom’s complex plant matrix. Current separation techniques, such as column chromatography, face significant difficulties in selectively isolating mitragynine, particularly given the diverse array of compounds present in kratom. Therefore, there is an urgent need for more efficient and selective methods for isolating mitragynine that can address the limitations of traditional techniques.
One technique for isolating mitragynine is column chromatography. This approach is an efficient technique for the separation and purification of mixtures. 4 In column chromatography, the choice of the stationary phase poses a problem for isolation, especially regarding its selectivity and compatibility with the sample. Consequently, the choice of the stationary phase poses a problem in attaining more efficient separation for the precise identification of secondary metabolites found in kratom. Molecularly imprinted polymers (MIPs) have garnered interest for their superior efficacy and selectivity in the separation of target compounds. Numerous prior studies have proven the application of MIP for the extraction of secondary metabolites from plants. This underscores the capability of imprinted polymers for the selective and precise isolation of target molecules. 5 The MIP designed to isolate plant metabolites has proven to be an effective method for identifying, extracting, and isolating compounds from plants. As a column adsorbent, MIP can be utilized to selectively separate and purify various compounds within the complex plant system.
Recent studies also show that MIPs have been effectively employed in the extraction of complex compounds from plants with challenging matrices, further demonstrating their potential in the isolation of secondary metabolites such as polyphenols. A novel strategy was developed for synthesizing functionalized silica monoliths as an artificial receptor for gallic acid using a sol-gel process. This method successfully extracted gallic acid from orange juice samples with high efficiency and selectivity, achieving excellent linear correlation for quantification. 6 Similarly, MIPs have been applied to the extraction of flavonoids, such as luteolin, demonstrating their ability to selectively bind and isolate these compounds with high adsorption capacity and selectivity. 7 Additionally, MIPs have been developed for the extraction of alkaloids. This MIP showed strong performance in separating scopolamine from a mixture of tropane alkaloids, achieving recovery rates of 96.0%–106.0% and demonstrating good linearity. The results confirm that MFMA-MIPs are highly effective for the selective isolation of scopolamine from complex alkaloid mixtures. 8 The current conditions promote the advancement of novel techniques for extracting secondary metabolites from kratom leaves. 9 However, despite the success of MIPs in isolating various plant metabolites, their application to the selective isolation of mitragynine from kratom has not been fully explored. While MIPs have demonstrated the ability to separate target compounds with high selectivity, the use of MIPs for mitragynine isolation remains under-investigated. This gap in the literature presents an opportunity to explore the efficacy of MIPs in isolating mitragynine from kratom leaf extract, providing a potentially more selective and efficient alternative to conventional chromatography. This study aims to address this gap by investigating the use of MIPs as selective sorbents for isolating mitragynine from kratom leaf extracts. The research will assess whether MIPs can offer superior results compared to traditional methods, in terms of both efficiency and selectivity. By providing a novel approach to the isolation of mitragynine, this study hopes to contribute valuable insights into the optimization of methods for isolating bioactive compounds from kratom, with potential applications in both scientific research and the pharmaceutical industry.
Materials and Methods
Materials
Chemicals
This study employed the following chemicals: colchicine, methacrylic acid (MAA), ethylene glycol dimethacrylate (EGDMA), azoisobutyronitrile (AIBN), kratom (M. speciosa Korth) leaf powder, methanol, n-hexane, ethyl acetate, 25% ammonia, purified water, cotton, sea sand, GF 254 TLC plates, HPTLC plates (20 × 10 cm), and Whatman 125 mm filter paper. All materials used were of analytical grade.
Instruments
This study employed a reflux apparatus, analytical balance, UV-Vis spectrophotometer (Shimadzu UV-Vis 1800), Fourier Transform Infrared Spectroscopy (FTIR) spectrophotometer (Shimadzu IR Tracer-100), High-Performance Thin-Layer Chromatography (HPTLC) apparatus (CAMAG), oven (Memmert), sonicator, rotary evaporator, centrifuge, vacuum pump, TLC chamber, capillary tubes, and chromatography column. Glassware and various standard laboratory instruments, including beakers, graduated cylinders, stirring rods, spatulas, droppers, mortars and pestles, and micropipettes, were also utilized in the experiments.
Selection of Dummy Template
A structural similarity study was carried out utilizing software tools, including ChemmineTools, QSAR, and OpenBabel, because of the limitations in the utilization of mitragynine. The tools were used to compare the data on alternative compounds with mitragynine after evaluating using the structural diagrams or Simplified Molecular-Input Line-Entry System (SMILES) notation. The Tanimoto coefficient was used to assess the structural similarity, with values close to one signifying greater similarity.
Prepolymerization Study
Prior to the polymer synthesis process, an investigation of the association constant and stoichiometric reaction was conducted to examine the interaction between the template and MAA as a functional monomer (FM) to form a complex. The association constant (Ka) was calculated through the UV titration method, commencing with the introduction of 1 mL of FM solution into the pre-homogenized template molecules within the solvent. Absorbance was subsequently monitored as the concentration of the FM increased, with observations recorded at the maximal wavelength of colchicine, 243.5 nm. The resulting data were graphed to illustrate the variation in absorbance (ΔA) against the concentration of the FM, thereby establishing their association through the Benesi-Hildebrand method. This was followed by Job plot analysis to determine the molar fraction of the template molecule and elucidate the stoichiometry of the complex formed between the template molecule and the FM. A colchicine solution (8 mg/L) and a monomer MA solution (0.1 mg/L) were prepared with different molar fraction ratios. Each ratio was pipetted into test tubes and subsequently homogenized. The total volume of the colchicine and monomer MA mixture was 4 mL. Subsequent to the preparation of these ratio variations, the mixture was analyzed by UV-Vis spectrophotometry and processed to determine the optimal synthesis ratio of template and monomer for further MIP synthesis.
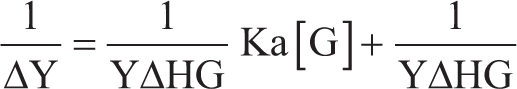
ΔY: Change in absorbance.
YΔHG: Change in absorbance with absorbance at the endpoint of the titration.
Ka: Association constant.
[G]: Concentration of the added monomer.
Synthesis of MIP
Optimization Synthesis
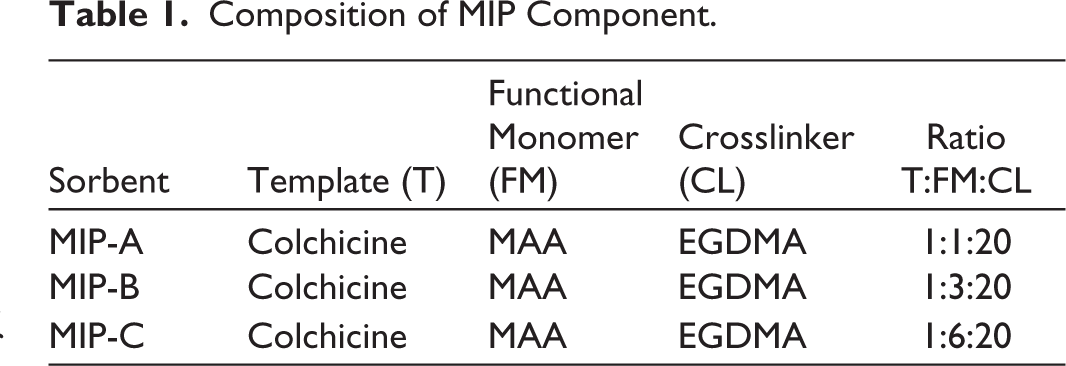
Bulk polymerization was employed for MIP synthesis. Three MIP ratios for this study were performed as shown in Table 1. The template molecule was dissolved in methanol with MAA monomer in a bottle, then it was combined with 3.94 mL of EGDMA and sonicated for 5 minutes. 0.3 mL of AIBN was added to the solution, and then nitrogen gas was run through the bottle for around 5 minutes. The oven was set to 80°C for 24 hours to heat up and perform the MIP polymerization. The polymer became a solid powder after being synthesized in the oven and then ground with a mortar and pestle until it formed fine particles. To find out the MIP yield, the solvent was evaporated, and the fine polymer was weighed. Optimization to obtain the maximal MIP yield was performed on the AIBN volume, temperature, and polymerization time. AIBN was added in different amounts, ranging from 0.05 to 0.5 mL, along with additional synthesis components. The temperature was optimized at 60, 70, and 80°C, and polymerization times of 6, 12, 24, and 48 hours were optimized. The optimal parameter was determined by analyzing the highest yield. Additionally, a non-imprinted polymer (NIP) was synthesized using the same method as MIP without a template for MIP comparison.
Composition of MIP Component.
Template Removal
The sonication method was used to evaluate the success of template removal from MIP. The MIP and NIP sorbents will be placed in a bottle containing 50 mL of methanol. The container will then be sonicated for 30 minutes. Subsequently, the sorbent will be filtered using a Buchner funnel lined with filter paper. The MIP or NIP sorbent that passes through the filter paper will be transferred into a tube for centrifugation. The supernatant will then be collected using a micropipette and analyzed using UV-Vis spectrophotometry at the maximum absorption wavelength of colchicine (243.5 nm), and the absorbance value will be observed. This process will be repeated until the colchicine peak is no longer detected, indicated by no absorbance value of colchicine observed at its maximum wavelength. The procedure was performed for a total of 16 cycles.
MIP Characterization
The MIP sorbents before and after template removal were analyzed using FTIR. The samples were placed in an Attenuated Total Reflectance (ATR) sample holder and pressed using a clamp to ensure proper contact between the sample and the crystal. The samples were then analyzed in the wave number range of 4,000 to 400 cm⁻ 1 . The colchicine standard was also analyzed as a reference.
Adsorption Study
Optimization Condition
Samples of MIP and NIP, with weights of 25, 50, and 100 mg, were placed in separate vials. Then, 4 mL of colchicine solution at a concentration of 8 mg/L was added to each vial. The mixtures were agitated using a shaker at 150 RPM for 60 minutes at room temperature, followed by filtration. Subsequently, colchicine was analyzed using a UV spectrophotometer at the maximum wavelength of 243.5 nm. The amount of compound adsorbed per gram of MIP and NIP was determined. The adsorption test also allows the determination of the imprinting factor (IF), which reflects the binding strength of MIP compared to NIP.
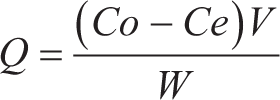
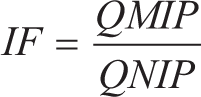
Q: Amount of substance adsorbed by MIP/NIP (mg/g)
Co: Initial concentration (mg/L)
Ce: Final concentration (mg/L)
V: Sample volume (L)
W: Weight of MIP/NIP (g)
Dynamic Adsorption
A 25 mg sample of MIP and NIP was placed in a vial, followed by the addition of a colchicine solution with a concentration of 8 mg/L. The vial was then agitated using a shaker at a speed of 150 RPM for different time intervals: 30 minutes, 60 minutes, 120 minutes, 180 minutes, and 360 minutes, followed by filtration. Subsequently, colchicine was analyzed using a UV spectrophotometer at the maximum wavelength of 243.5 nm. The amount of compound adsorbed per gram of MIP and NIP was determined.
Static Adsorption
A 25 mg sample of MIP and NIP was placed in a vial, to which 4 mL of colchicine solution with varying concentrations (2, 3, 4, 5, 6, 7, and 8 mg/L) was added. The vial was then shaken at 150 RPM for 120 minutes, followed by filtration. Subsequently, colchicine was analyzed using a UV spectrophotometer at the maximum wavelength of 243.5 nm. The amount of compound adsorbed per gram of MIP and NIP was determined.
Kratom Plant Preparation
The kratom plants were purchased from a vendor. Before being used as a sample, kratom leaves will be sent to the Yayasan Generasi Biologi Indonesia for species determination. For the extraction process, 15 grams of kratom leaves were placed into a 500 mL round-bottom flask, followed by the addition of 300 mL of methanol as the solvent, ensuring the leaves are completely submerged. The round-bottom flask was then attached to a reflux apparatus that had been pre-set up. Extraction was carried out for 2 hours at a temperature of 60°C, after which the extract was cooled. Subsequently, filtration was performed using Whatman 125 mm filter paper to separate the filtrate from the residue. The extract was concentrated using a rotary evaporator at 50°C, with a rotation speed of 120 RPM and a pressure of 0.06 to 0.07 MPa for approximately 90 minutes. The extract was then transferred to an evaporation dish and covered with perforated aluminum foil, allowing it to rest at room temperature for the complete evaporation of methanol. The weight of the extract was measured, and the extract was stored in a closed container. The yield from the extract was calculated.
Screening of Mitragynine in the Extract
A silica gel 60 F254 TLC plate (8 × 10 cm) was prepared, and 1 cm lines from the bottom and a 0.5 cm line from the top were marked on the plate. The plate was then placed in an oven at 100°C for an hour to activate it. The plate was allowed to cool to room temperature once it was activated. Subsequently, the plate was put under UV light at 254 nm and 366 nm to make sure there were no impurities on it. A 5 mL combination of n-hexane, ethyl acetate, and 25% ammonia in a 30:15:1 (v/v/v) ratio was utilized as the mobile phase. The mobile phase was added to the chamber, sealed, and left to soak for 45 minutes. A paper that was dipped slightly into the chamber was used to keep an eye on the saturation of the mobile phase. Once the mobile phase rapidly rose up the plate, it indicated that the chamber was saturated.
Sample spotting was performed using the Linomat V instrument with the VisionCATS software. The mitragynine standard was spotted onto the plate at a volume of 1 µL. The kratom extract was dissolved in a small amount of the mobile phase, and the mobile phase containing the extract was then spotted at 2 µL. The plate was then visualized under UV light at wavelengths of 254 nm and 366 nm. The TLC plate was placed into a pre-saturated chamber, ensuring the spotted area was positioned at the bottom. The chamber was sealed, and the plate was developed until the solvent front reached the desired position. After development, the plate was taken out and allowed to dry. The entire plate was then visualized again under UV light at wavelengths of 254 nm and 366 nm to observe the separation results. After development, the plate was examined under UV light at wavelengths of 254 nm and 366 nm, and the Rf values were calculated. Mitragynine will appear on the plate under 366 nm UV light as a bright green spot, with an Rf value similar to that of the mitragynine standard.
Separation of Mitragynine
Column Preparation and Fraction Separation
Thirty vials were prepared for calibration; the outside of each vial was wiped out using ethanol and a cloth. 15 mL of ethanol was measured using a graduated cylinder and added to each vial. A label showed how much ethanol was contained in each vial. The same method was used to calibrate all of the vials. A long cylindrical glass column was prepared and rinsed with 100 mL of water, followed by 50 mL of a mixture of n-hexane:ethyl acetate:25% ammonia in a 30:15:1 ratio. The column was then clamped and stood, with the bottom facing down so it could dry. The column was put back in its original place after it dried, and the column stopcock was closed. A piece of cotton was put at the top of the column’s neck, soaked with solvent, and squeezed down until the neck was completely closed with no air trapped inside. 2,5 grams of sea sand were weighed out and slowly added to the column while adding a little bit of solvent to it. The solvent was poured into the column until the sand was completely covered MIP sorbent was placed in an oven at 80°C for 30 minutes before use. A total of 5 grams of the MIP was taken as the stationary phase, and the remaining 250 mg were used for preadsorption of the extract. The 5 grams of NIP were placed in a beaker, wetted with the solvent mixture (n-hexane:ethyl acetate:25% ammonia, 30:15:1 ratio), and then transferred to the column. The slurry and solvent were continuously poured, and the beaker was rinsed until the entire stationary phase was evenly distributed in the column. The surface of the NIP was leveled using a stirring rod. Then, the column was eluted with a mix of n-hexane, ethyl acetate, and 25% ammonia in a 30:15:1 ratio. Every 15 mL of the fractions that came out of the column was put into separate vials, labeled, and kept. Using TLC and HPTLC, each fraction was then checked for separation patterns and the presence of mitragynine.
A 10 × 20 cm silica GF254 plate was prepared and then heated in an oven at 100°C for an hour to activate it. The mobile phase used was a 75 mL mixture of n-hexane, ethyl acetate, and 25% ammonia in a 30:15:1 ratio. The chamber was filled with the mobile phase, and vaseline was put on the top and lid of the chamber. The chamber was closed and filled for 45 minutes. The standard, extract, and fraction samples from the separation were prepared on the plate: 1 µL for the standard and 2 µL for the extract and fractions. The plate was placed in the chamber for elution, then the plate was taken off, dried, and observed. Finally, the CAMAG TLC scanner was used to analyze the separation patterns on the TLC plate and to determine the area under the curve (AUC) for each separation band.
Quantification of Mitragynine and Isolation Efficiency
The AUC of each fraction was compared to that of mitragynine in the full extract in order to determine the amount of mitragynine present in each fraction. The total mitragynine recovered from the separation was then determined by estimating the mitragynine content in the fractions using this comparison. The isolation efficiency was determined by comparing the expected mitragynine content in the fractions with the theoretical amount, which was based on the average mitragynine concentration in the kratom extract as documented in the literature. The efficiency of the isolation process was also compared to that of the standard mitragynine purchased from the vendor.
Isolate Characterization
The obtained isolate was compared with the mitragynine standard using FTIR. The samples were placed in an ATR sample holder and pressed using a clamp to ensure proper contact between the sample and the crystal. The samples were then analyzed in the wave number range of 4,000 to 400 cm–1. Structural similarity between the isolate and the standard was calculated using R software (version 4.5.1). 10
Comparison With Other Methods
To highlight the advantages of MIPs over conventional techniques, which often suffer from low selectivity and extended processing times, a comparison with preparative thin-layer chromatography (PTLC) was performed. Following the activation process for an hour at 100°C in an oven, the GF-254 silica gel plate (20 × 10 cm) was allowed to cool before being examined under a UV lamp. After the mobile phase optimization process, the optimal mobile phase was identified and diluted to a 50 mL volume. The mitragynine standard was spotted as a reference, and a 128 µL sample of the methanol extract was spotted onto the plate using a Linomat V applicator to guarantee precise volume and sample distribution. Various parameters, including saturation time, plate pre-conditioning time, migration distance, and plate drying time, were optimized. To prepare the mobile phase, 10 mL of pre-conditioning solvent and 25 mL of developing solvent were used. The HPTLC plate, which had been spotted, was then placed into the ADC for pre-drying to ensure the spots were dry. To saturate the chamber, it was preconditioned for 15 minutes. Seven centimeters was the elution distance used. The plate was taken out and allowed to dry for 5 minutes following the elution process. The eluted spots were then visualized under UV light at wavelengths of 254 nm and 366 nm. Spots in the sample that aligned with the mitragynine standard were scraped. The spots that had an Rf value identical to the mitragynine standard were carefully scraped off, dissolved in methanol, and centrifuged to separate the silica gel particles. The supernatant was collected as the crude mitragynine isolate. The purity of the isolate was evaluated using two-dimensional TLC. The yield of the isolate was calculated by comparing the weight of the purified isolate to the total weight of the original sample in order to evaluate the efficiency of the isolation procedure.
Results and Discussion
Selection of Dummy Template
The limited availability of mitragynine as a template molecule posed a significant challenge for this investigation, necessitating the exploration of other molecules as alternatives. In most MIP studies, the template molecule is the target compound to be isolated. However, when the target molecule cannot be used as the template, a dummy template is an appropriate alternative. The use of a dummy template allows for the replication of the target molecule’s structure, facilitating the formation of MIPs that are selective for the desired molecule. This method helps in optimizing the polymer’s binding sites, which in turn enhances the interaction between the analyte and the MIP during the isolation process, making it more efficient and selective. 11 The optimization of the dummy mitragynine template was carried out by computational methods with applications ChemMineTools, OpenBabel, and QSAR. These three software programs were chosen because they are easy to use and work quickly. The Tanimoto coefficient measures how similar two chemical substances are by looking at their structures. SMILES, ChEMBL, and fingerprints from PubChem were used for the structural similarity study. The Tanimoto coefficient was used to quantify this similarity, with a coefficient near one indicating a high degree of structural similarity between the compounds. 12 Based on the method, colchicine was found to be a promising substitute for mitragynine because of its availability in the laboratory, with a structural similarity of 46.49%. Both compounds possess a similar number of hydrogen bonds; mitragynine has seven (Figure 1) hydrogen bonds, whereas colchicine has seven. Colchicine is soluble in the organic solvent methanol, and has a molecular weight similar to that of mitragynine (399.4 g/mol for colchicine vs. 398.5 g/mol for mitragynine).13,14 Therefore, colchicine was selected as the dummy template for the further laboratory study.
Chemical Structures of Mitragynine (a) and Colchicine (b).
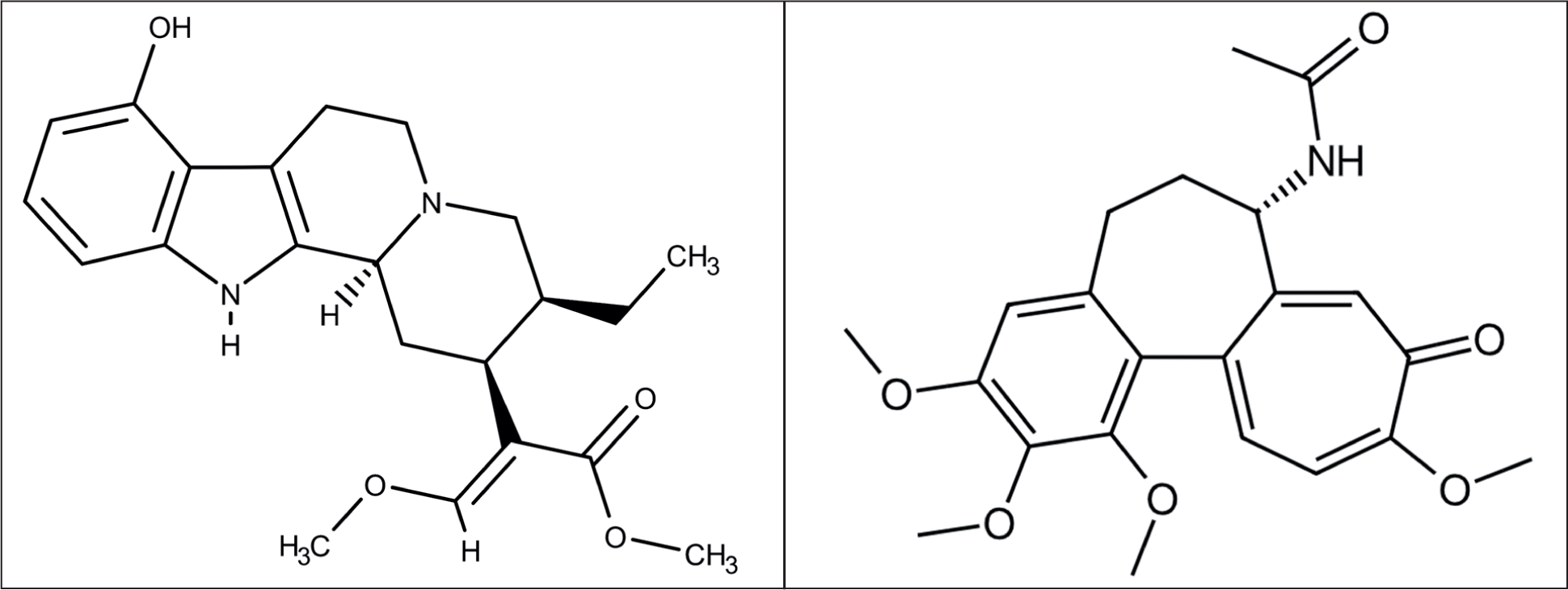
Prepolymerization Study
There are five main parts of MIP synthesis: the template molecule, the FM, the crosslinker (CL), the porogenic solvent, and the initiator polymerization reaction. The interaction between the template and FM is crucial, as it significantly affects the binding affinity of the MIP. 15 Before carrying out the synthesis study, a prepolymerization examination was performed to analyze the interaction between colchicine as the template and MAA as the FM. The selection of FMs is a crucial determinant affecting the selectivity and efficacy of MIPs. The functional groups of the monomers interact with the template molecule during the prepolymerization phase, forming complexes which establish the properties of the MIP. These interactions significantly impact the overall characteristics of the MIP, encompassing its binding capacity and selectivity for the target molecule. A more reliable interaction between the template and the monomer increases the bonding capacity and selectivity of the MIP. 16 MAA was chosen as the monomer due to its suitability and availability in the laboratory.
The carboxylic acid group of MAA (-COOH) interacts with functional groups on colchicine, primarily through hydrogen bonding. Specifically, the -OH of MAA acts as a hydrogen donor to acceptor sites on colchicine, such as the carbonyl groups in the structure. At the same time, the C=O of MAA can accept hydrogen from colchicine’s NH. This is facilitated by the rigid structure of colchicine, minimizing steric hindrance. These interactions, enabling the formation of hydrogen bonds, drive the complex formation with the hydroxyl group of MAA. 17 Non-covalent interactions, particularly hydrogen bonding between colchicine and MAA, play a vital role in the synthesis. The hydrogen bonds contribute to the formation of a stable MIP, which is defined by a strong and reversible contact between the template molecule and the monomer. This characteristic makes it easier to make the MIP form, which makes it less challenging for the template removal process.
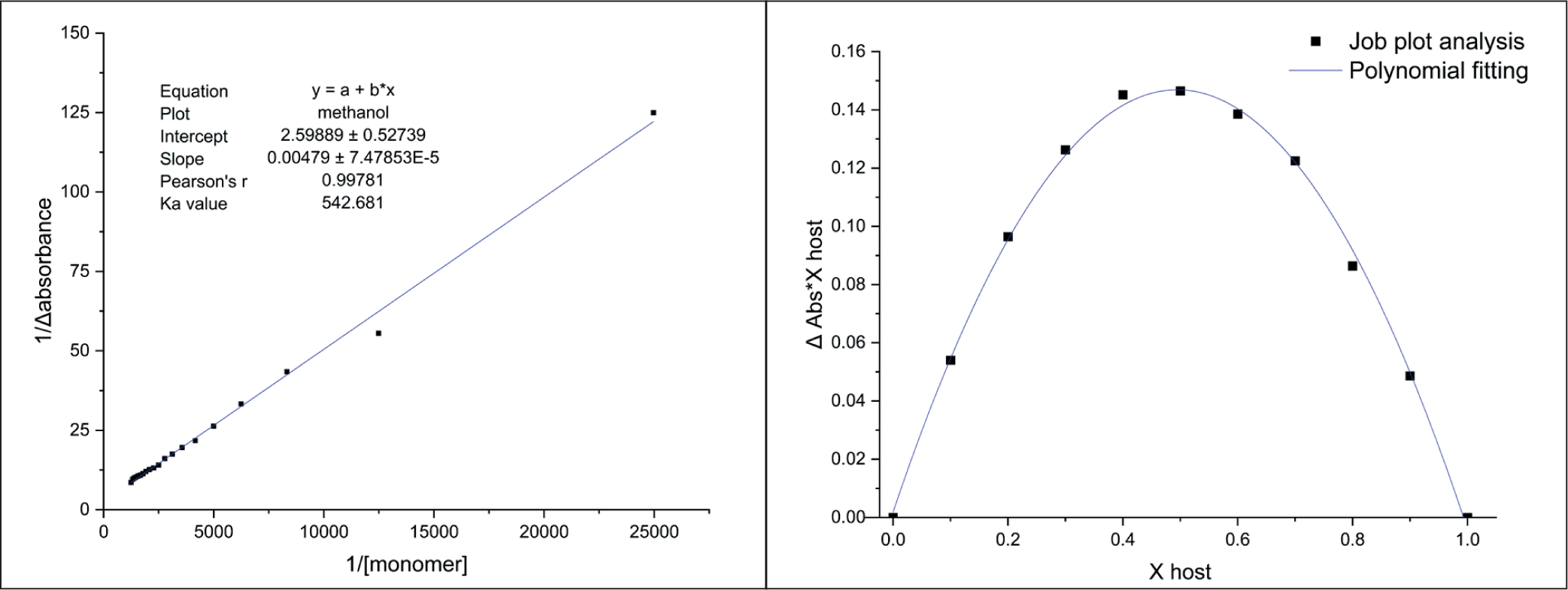
The association constant (Ka), which quantifies the strength of the interaction between template molecules and the chosen FM, was determined using a UV-Vis spectrophotometer at the maximum wavelength of colchicine absorption (243.5 nm). Molar equivalence between the template molecule and the FM was required for interaction, which was reflected in a decrease in absorbance. At this stage, 20 drops of the monomer were gradually added to the template molecule, and the absorbance was measured, which was then used to calculate the Ka value. The template molecule was analyzed in triplicate, all with the same concentration of 8 mg/L. UV-Vis spectrophotometry provided the absorbance data, and a graph was made to show how 1/monomer concentration (x-axis) and 1/Δ absorbance (y-axis) are related in order to find the Ka value. The Ka value has a wide range; it is under 25 M−1 for weak intermolecular interactions and surpasses 100 M−1 for strong intermolecular interactions,18,19 Colchicine exhibited Ka values of 481.9 ± 52.8 M⁻ 1 , which are consistently above 100 M⁻ 1 , indicating a strong interaction between the template molecule and the monomer. Therefore, the MIP can recognize the colchicine binding site and form the complex in the structure. 20
The stoichiometry of the process was figured out to determine the optimal template-to-FM ratio in the prepolymerization complex. They are a critical tool in the rational design of MIP. They are used to determine the optimal molar ratio between a template molecule (target) and the FM(s) that will create the most stable prepolymerization complex, ensuring the resulting polymer has high affinity and selectivity. 21 Excess monomer can lead to less selective binding sites, making it harder to isolate the desired analyte. However, if an adequate amount of monomer is not utilized, polymerization may not occur at all. The optimal ratio of monomer to template is considered highly critical for ensuring that the appropriate binding sites are formed during MIP synthesis. Job plot analysis was employed to accomplish the stoichiometric ratio in the investigation. 22 The optimal molar ratio is indicated by a y value of 0.1465, as shown in Figure 2. It is suggested that a y/x ratio close to three should be present for the formation of the most stable equilibrium complex. It has been demonstrated that maximum stability is achieved at a 1:3 stoichiometric ratio in the host-guest interaction between mitragynine (template) and MAA (monomer). This ratio served as the reference template for the monomer ratio for further MIP synthesis.
UV Titration (Left) and Job Plot (Right) of Colchicine-MAA.
Synthesis of MIP
Optimization Synthesis
The bulk polymerization method was used to synthesize MIPs because it is a well-established method that is relatively simple. In this method, the template molecule was combined with the FM, CL, and porogenic solvent. Then, an initiator was added to start the polymerization reaction. The mixture was subjected to controlled heating to form a rigid polymer matrix with binding sites that specifically match the template molecule. The formula for the synthesis had a ratio of 1:3:20 for colchicine:MAA:EGDMA. For the CL, EGDMA was chosen, and for the initiator, AIBN was chosen since both of them are commonly used in the MIP synthesis. After that, the polymers were ground and sifted through a 60-mesh screen to ensure uniform particle size. The bulk synthesis method used in this study resulted in polymer powders with particles of different sizes. The irregular shapes can change how colchicine interacts with the binding sites in the MIP, which can lead to non-specific binding. Therefore, gradual grinding was done to obtain uniform polymer sizes, making sure that the template molecule release process was not affected. 23 Bulk polymerization was chosen primarily for its procedural simplicity and the absence of complex stabilizing agents. Unlike alternative methods such as emulsion or suspension polymerization, bulk synthesis does not require surfactants or stabilizers that could potentially compete with the template-monomer interactions. This ensured that the integrity of the prepolymerization complex was maintained, allowing for a direct evaluation of the imprinting efficiency. This approach allowed for a straightforward scaling of the synthesis to produce sufficient material for both batch adsorption studies, without the complexities of multi-step parameters. 24
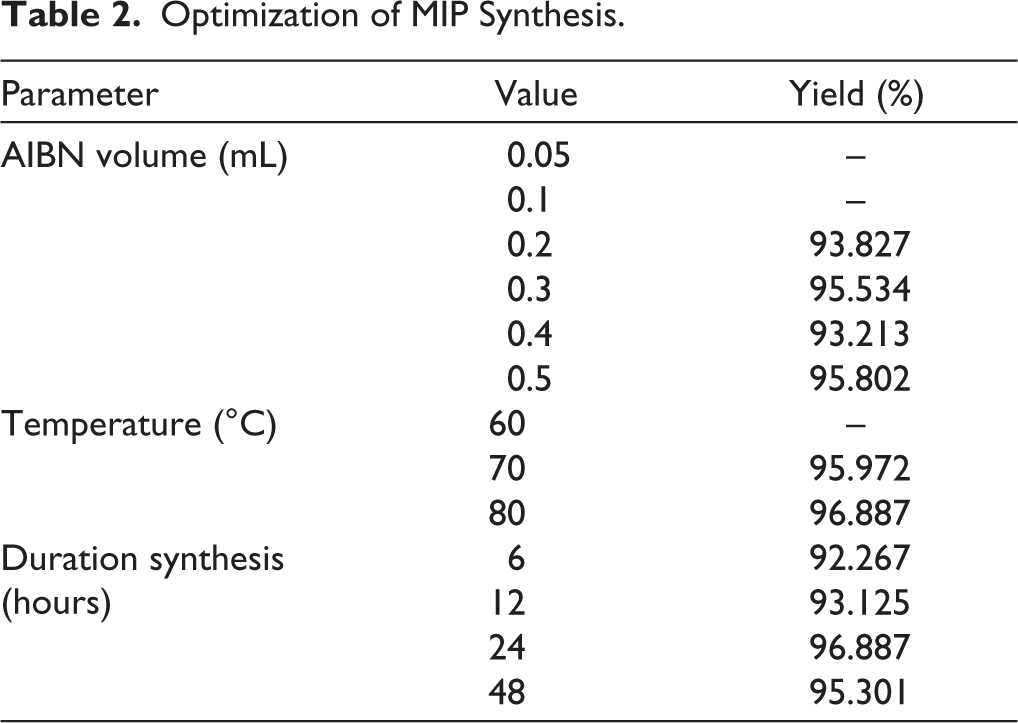
The optimization stage was crucial to ensure that the synthesized MIP had the proper characteristics for selective adsorption and separation. The concentration of the initiator, the temperature of the polymerization, and the reaction time were all investigated carefully, and the corresponding optimization yields are summarized in Table 2. These are crucial since they all have significant impacts on how effectively the polymer forms and how stable the MIP becomes. The amount of AIBN was identified as a crucial determinant: at low volumes (0.05–0.1 mL), polymerization failed to occur in place, whereas 0.2–0.5 mL provided substantial results. The best result came from using 0.3 mL of AIBN, which made polymers rapidly without utilizing an excessive amount of initiator. The temperature also had an effect on the process. No polymer was formed at 60°C, but the amount of polymer increased significantly at 70°C and reached its highest point at 80°C. This means that higher thermal energy makes radical initiation more likely, but temperatures above 80°C could damage the material. A similar trend was observed with reaction time. While shorter times gave relatively high yields, the most consistent and stable results were seen after 24 hours. Extending the time to 48 hours did not increase yield and may have caused damage to the produced MIP. In general, the best conditions for making MIP and NIP were found to be 0.3 mL AIBN at 80°C for 24 hours. This gave significant yields with excellent polymer quality for further adsorption studies.
Optimization of MIP Synthesis.
Template Removal
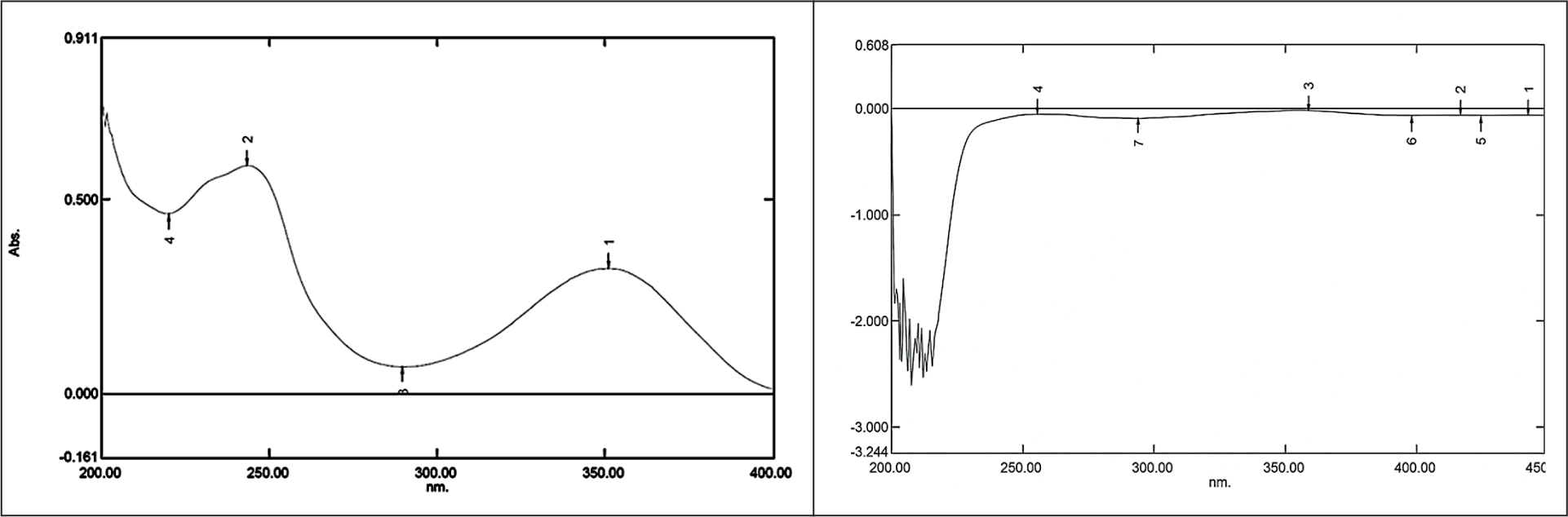
Template removal is important because it directly dictates the number of available recognition sites and the overall sensitivity of the MIP. Its effectiveness determines the accessibility of the imprinted cavities; incomplete removal not only reduces the maximum adsorption capacity but also leads to “template bleeding,” where residual molecules leach into the analyte solution and compromise the accuracy of subsequent measurements. The template molecule removal of colchicine was conducted using the sonication method. Sonication offers advantages in terms of efficiency, utilizes vibrations to release the template molecule, and produces satisfactory release results. 25 In the template removal process, MIP samples were sonicated with methanol for 30 minutes and then filtered using filter paper and a Buchner funnel to speed up the filtration process. If a peak was detected in the UV-Vis spectroscopy, it indicated that MIP still contained the colchicine as a template molecule and had not been fully removed from the sorbent. Colchicine was successfully released from the MIP during the 16th cycle, as indicated by the absence of the colchicine peak at wavelengths 243.5 nm (Figure 3).
UV Spectrum of Colchicine Standard (Left) and MIP After 16th Cycle (Right).
MIP Characterization
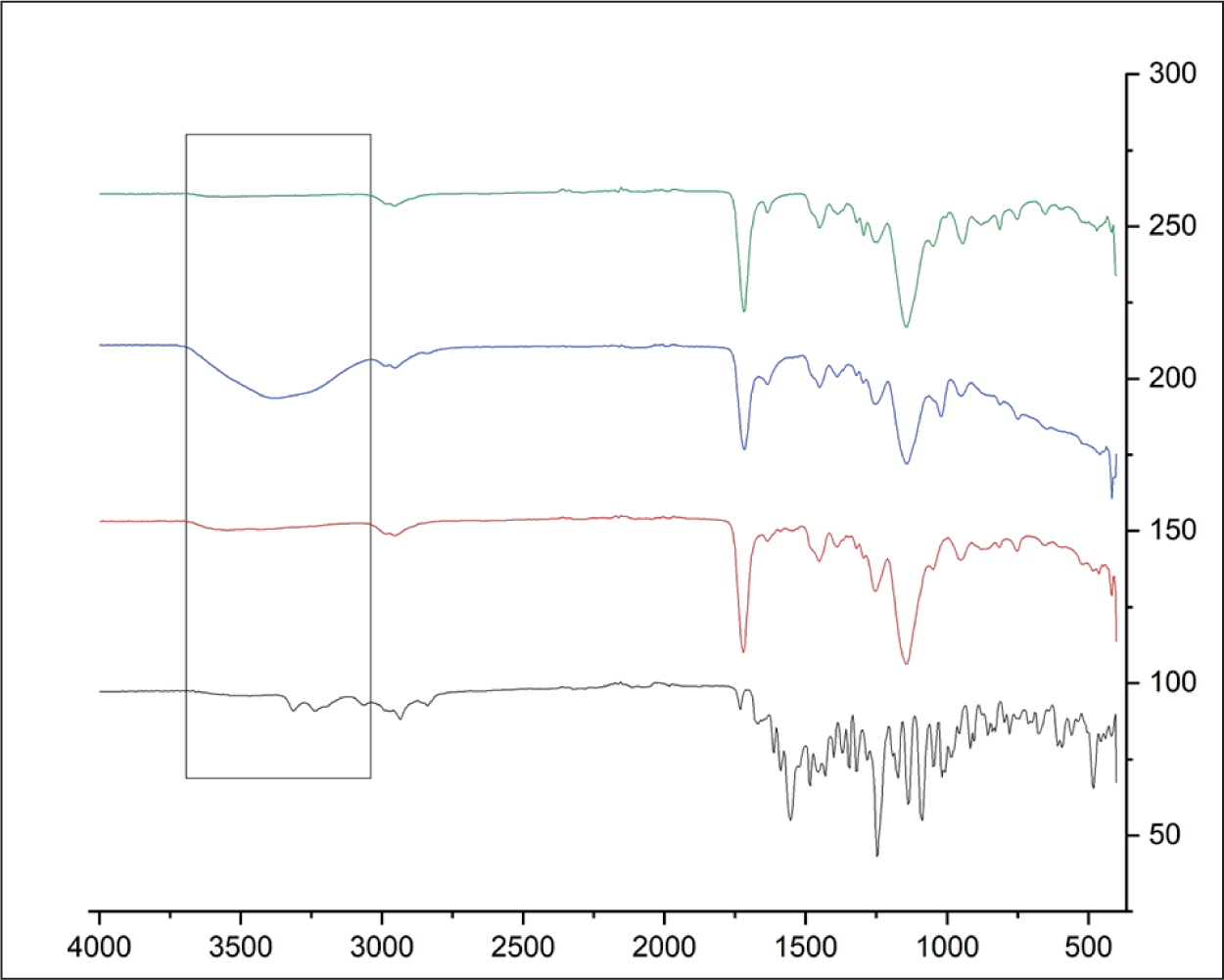
The structural, morphological, and chemical properties of the synthesized MIP and NIP were comprehensively characterized to evaluate the success of the imprinting process. FTIR was used to identify functional groups present in the MIP and to confirm the successful formation of the polymer. The IR spectra were also used to make sure that the template molecule had been removed successfully from the synthesized MIP. 26 FTIR analysis was performed on several samples: colchicine as the template molecule, the MIP before template removal, the MIP after template removal, and the NIP for comparison. Figure 4 shows the overlay IR spectra for these samples. The colchicine standard exhibited that there were several functional groups, like the NH2 group at 3,350–3,180 cm⁻ 1 and the CH group at 2,870–2,800 cm⁻ 1 . In the MIP before the template was released, the functional groups like the CH group at 2,870–2,800 cm⁻ 1 and the C=O carboxyl group at 1,730–1,700 cm⁻ 1 were observed. The OH group at 3,400–3,200 cm⁻ 1 was the most important functional group after the template was released. Before the template was removed, the -OH band could not be seen in the MIP. After the template removal process, the OH band at 3,400–3,200 cm⁻ 1 became clearly visible in the MIP spectrum, confirming the successful release of colchicine from the polymer. The appearance of this hydroxyl group band post-template removal suggests that colchicine had interacted with the -OH groups of MAA during the polymerization process, and the template was successfully extracted. This observation confirms that colchicine was successfully removed from the MIP sorbent, ensuring that the MIP is now capable of selectively binding to future template molecules with similar structural characteristics. 27
IR Spectrum of Colchicine (Black), MIP Before (Red) and After (Blue) Removal, NIP (Green).
Adsorption Study
Optimization Condition
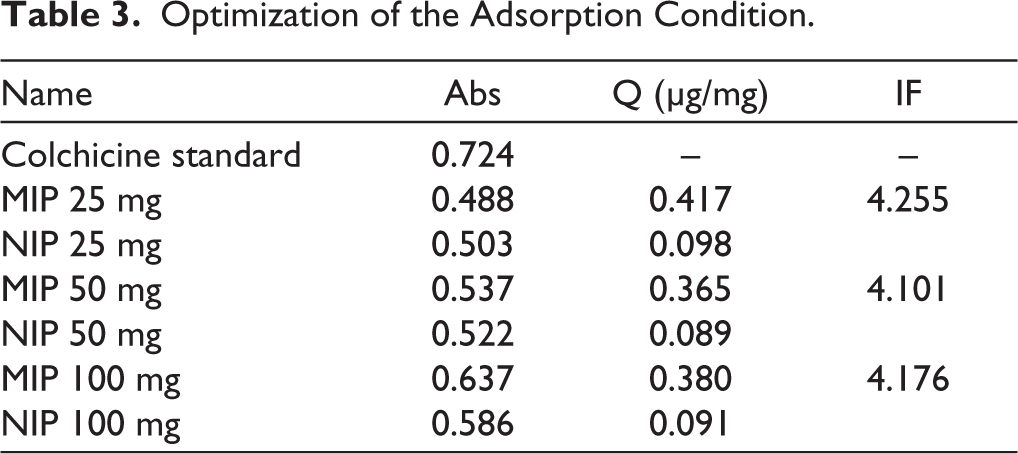
Adsorption optimization was carried out to determine the most effective amount of MIP required for adsorbing the colchicine after template removal. The amounts of MIP tested were 25, 50, and 100 mg, and the adsorption performance was evaluated based on the adsorption capacity (Q) and the IF. As shown in Table 3, the 25 mg MIP sample produced the highest Q value (0.417 µg/mg) with an IF of 4.255. Raising the MIP mass to 50 mg and 100 mg did not make adsorption better; instead, the Q values went down slightly, while the IF values stayed in a similar range. Adding more MIP sorbent should help capture the analyte because there will be more binding sites available. But the fact that Q values went down at higher polymer masses suggests that other factors may have affected the adsorption process. When excessive amounts of MIP are used, particle aggregation may occur, thereby reducing the effective surface area available for binding. Diffusion of the analyte into the binding sites was hindered under conditions with larger polymer quantities, resulting in less effective utilization of the sorbent.
Optimization of the Adsorption Condition.
Dynamic Adsorption
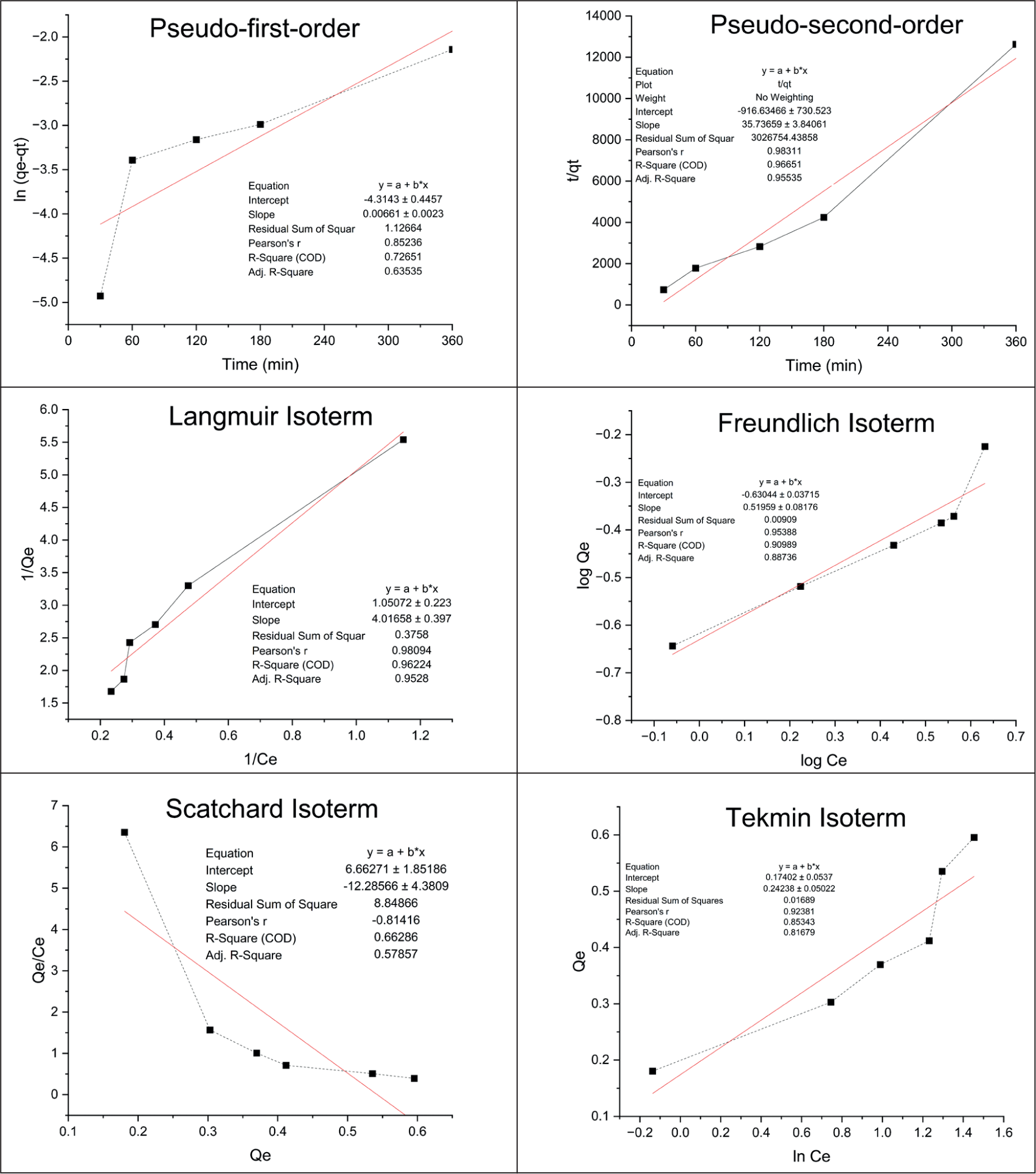
The dynamic adsorption test was conducted to determine the rate of colchicine adsorption by MIP at specific time intervals. This experiment was designed to determine how colchicine interacts with the surface of the MIP, providing insight into the adsorption kinetics, the rate and extent to which colchicine binds to the polymer. This investigation is based on the amount of colchicine trapped on the surface of the MIP. The dynamic adsorption test is used to describe the relationship between the adsorbate and adsorbent, which is also referred to as adsorption kinetics. The adsorbate is the substance that is to be captured by the MIP; in this study, colchicine is the adsorbate, while the adsorbent is the surface on which the adsorbate attaches, in this case, the MIP. The adsorption kinetics can typically be described by two main models: pseudo-first-order and pseudo-second-order. The pseudo-first-order model assumes that adsorption occurs on only one side of the MIP surface, where the rate of adsorption is proportional to the concentration of free adsorbate. In contrast, the pseudo-second-order model represents adsorption that binds to specific binding sites on the MIP surface, suggesting that the adsorption process is governed by the interaction of the adsorbate with specific target sites on the adsorbent. 28 This test was carried out with different time variations, using the ideal colchicine concentration of 8 mg/L. The purpose of this test was to determine whether the MIP could capture or adsorb colchicine. The results of the dynamic test showed that the MIP exhibited optimal colchicine adsorption at 180 minutes. This is further illustrated in Figure 5, where the colchicine dynamic test follows a pseudo-second-order model with an r value of 0.983. 29 The pseudo-second-order model is typically more applicable for systems where specific interactions occur between the adsorbate and adsorbent, which is often the case with MIPs due to their highly selective binding sites. This demonstrates the ability of the MIP to selectively bind colchicine and suggests that similar MIP-based approaches could be used for the isolation process from complex mixtures.
Static Adsorption
The static adsorption test was performed to analyze the capacity of MIP to selectively adsorb colchicine as the template molecule. The test was performed with varying colchicine concentrations, ranging from 2 to 8 mg/L, and the samples were mixed for 180 minutes. The findings indicated that the peak Q value occurred at a concentration of 7 mg/L, indicating that this concentration was optimal for achieving the highest binding efficiency within the tested range. The Langmuir isotherm assumes that adsorption occurs through the formation of a monolayer on the surface of the adsorbent, with no interaction between adsorbed molecules. This model is most suitable when the adsorbent has specific binding sites, such as those formed in MIPs. According to this model, the peak adsorption capacity is reached when all binding sites on the MIP are occupied. The Freundlich isotherm, in contrast, suggests that adsorption occurs in multiple layers and is physically based, meaning the interaction between the adsorbent and adsorbate is relatively weak. The Temkin isotherm posits that the adsorption energy decreases linearly as the surface coverage increases, indicating a gradual decrease in binding strength. Finally, the Scatchard isotherm provides a measure of the dissociation constant (Kd), which quantifies the binding strength between the template molecule (colchicine) and the MIP. A higher Kd value indicates a stronger affinity between colchicine and the MIP. 30 Figure 5 shows that the Langmuir isotherm provided the best fit to the experimental data, with an R value close to 1 (R = 0.9809). The analysis indicates that colchicine exclusively binds to the designated binding sites, exhibiting no interaction with alternative binding sites. In the Langmuir isotherm, Qe represents the amount of adsorbate adsorbed, Qmax indicates the capacity of the monolayer adsorbent, Ce represents the adsorbate concentration at equilibrium, and KL is the Langmuir adsorption constant. An important parameter derived from the Langmuir isotherm is the separation factor (RL), which provides insight into the efficiency of the adsorption process. The RL value is calculated to assess whether the adsorption process is favorable (0 < RL < 1), unfavorable (RL > 1), or linear (RL = 1). In this study, the RL value was found to be 0.323, indicating that the adsorption process is effective, with favorable adsorption conditions. According to established guidelines, an RL value between 0 and 1 is considered optimal, suggesting that colchicine interacts efficiently with the MIP, making this system highly suitable for selective adsorption. The results from the static adsorption study demonstrate that the MIP has a high affinity for colchicine, confirming its potential as a selective sorbent for isolating mitragynine. 31
Dynamic and Static Adsorption of MIP.
Kratom Plant Preparation
The taxonomic determination confirmed that the examined samples were kratom leaves (M. speciosa Korth.), as shown in Supplementary Materials 1. The extraction process employed the reflux method for kratom leaf extraction. A prior study discovered that reflux was the best method for extracting data since it consistently gave a greater extract recovery, between 23.59% and 25.53%. 32 The experiment employs organic solvents like methanol and ethanol to extract kratom leaves, since mitragynine dissolves well in both solvents, with around 10 mg/mL in methanol and 5 mg/mL in ethanol. 33 Previous research has shown that methanol is the best solvent for kratom extraction since it works better. Consequently, methanol was chosen for this work to guarantee optimal solubility of mitragynine and to achieve an elevated extract yield. 34 The extraction process in this study was performed in four replicates; the combined extracts were concentrated to produce 15.0136 g of thick extract, resulting in a final yield of 25.023%. When compared with the results reported by the previous study using the reflux method, the present findings are consistent. These results confirm that the reflux extraction process was effective, yielding extracts at the higher end of the expected range.
Screening of Mitragynine in the Extract
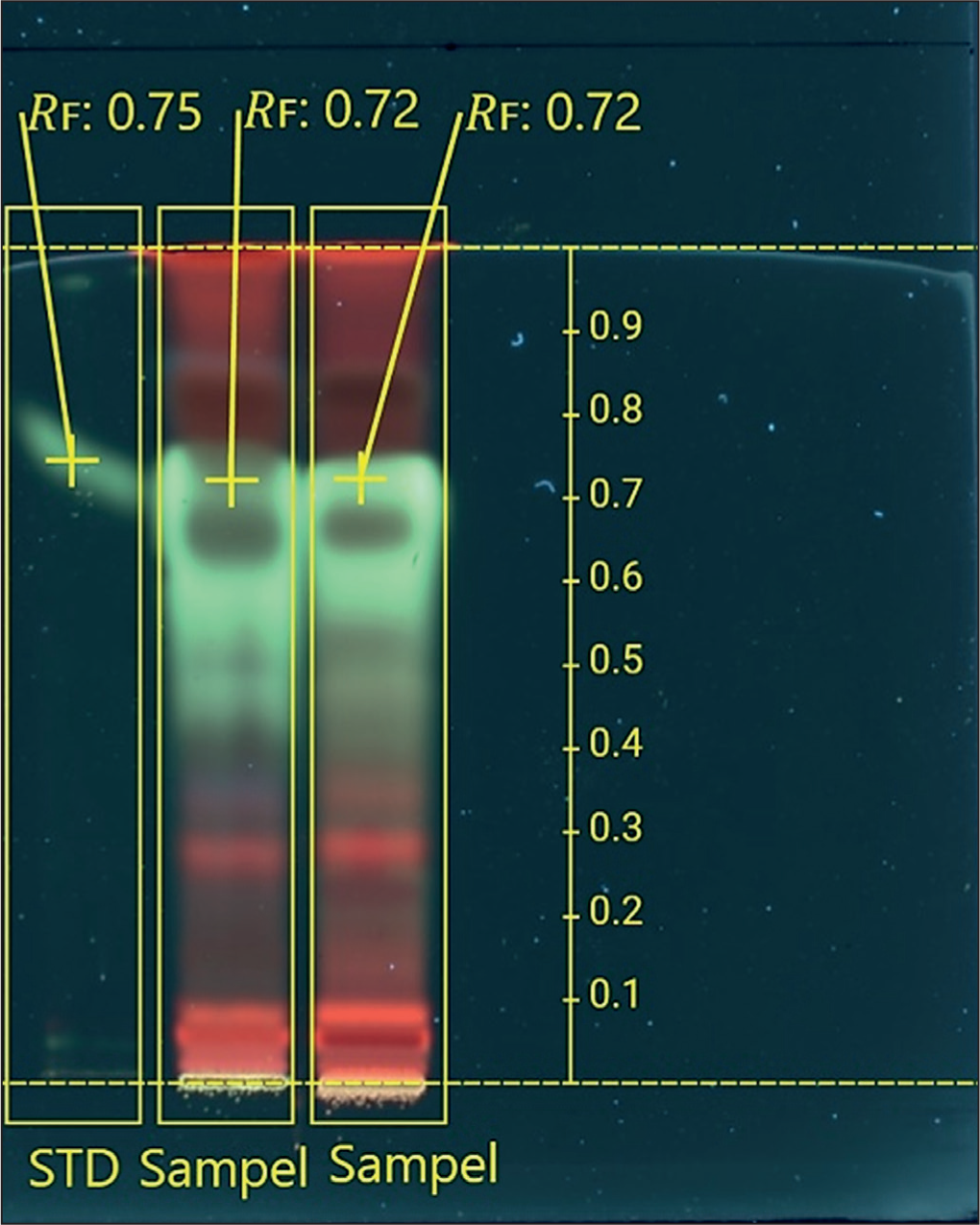
TLC was used to screen the extract by applying it to a silica gel plate and comparing its separation to that of a mitragynine standard. The mobile phase mixture was chosen because previous research using a different method demonstrated its effectiveness in separating mitragynine. 26 The visualization showed that the mitragynine standard had an Rf value of 0.75, while the spot in the extract had an Rf value of 0.72 under UV light at 366 nm (Figure 6). A spot of similar color appeared with a similar Rf value, indicating strong correspondence with the standard. The extract had more than just mitragynine; it also had other compounds that showed up as red spots at different Rf positions. The results indicate that the kratom leaf extract contained mitragynine, as evidenced by the presence of a compound exhibiting an identical color and Rf value to the standard. This step was necessary to make sure that the plant material used in the study was really kratom leaves that had mitragynine in them.
Mitragynine Screening TLC.
Separation of Mitragynine
Column Preparation and Fraction Separation
Two experiments were conducted for the variations in the quantity of sorbent, extract, and the chromatographic system applied in each experiment, as summarized in Table 4. In the second experiment, vacuum assistance was performed to accelerate the fraction collection process. In the first experiment, the separation was performed manually, requiring approximately 1 to 2 hours to obtain a single fraction. A total of 10 fractions were collected over a period of 3 days. The collection process was terminated at the tenth fraction because the chromatographic system became clogged, and no additional fractions could be eluted. Due to the relatively lengthy separation time, the subsequent experiment was conducted under vacuum-assisted conditions. This modification significantly improved efficiency, resulting in the collection of 30 fractions, with an average duration of 10–15 minutes per fraction.
Difference Condition in Column Separation.
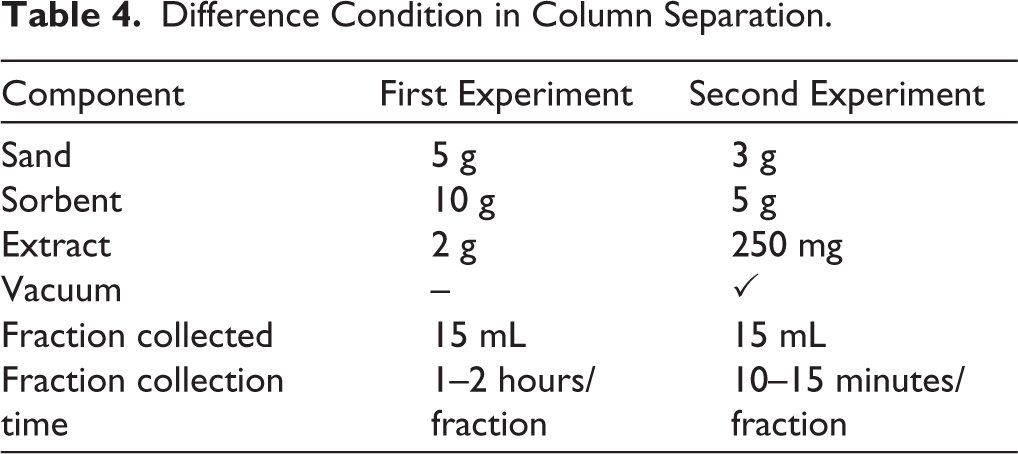
The MIP sorbent was utilized as the stationary phase in column chromatography to assess its viability as an alternative to conventional silica sorbent. MIP exhibits greater selectivity for certain target molecules compared to silica and its derivatives, which often lack selectivity or have limited selectivity. MIP preferentially interacts with target molecules. Silica, conversely, employs polar–nonpolar interactions or van der Waals forces. MIP demonstrates enhanced stability, permits prolonged reuse, and facilitates the modification of solvent conditions in accordance with the polymer formulation. Conversely, silica typically requires powerful organic solvents. MIP can be designed for specific target chemicals, offering it more versatile than conventional sorbents, which are limited to separations based solely on generic polarity. These advantages indicate that MIP may serve as an effective alternative to conventional silica sorbents. 35 The result demonstrated sufficient adsorption ability to separate mitragynine from other extract components, since the specific binding cavity characteristic was provided in the separation. This was confirmed by the separation patterns observed in the TLC results, which revealed that the MIP facilitated the earlier elution of extract components other than mitragynine in the first fraction. Mitragynine began to appear from the second fraction onward. By the sixth fraction, the separation pattern appeared cleaner, although traces of other compounds were still visible on the plate. Overall, the system had not yet reached full optimization, as indicated by the column becoming clogged before the elution process was complete. Despite this, mitragynine was still detected up to the tenth fraction, with separation bands that were not entirely separated. In the second experiment, the TLC results demonstrated that the sorbent provided better selectivity as an adsorbent. In the first fraction, mitragynine was observed to elute first, while most of the non-mitragynine extract components were retained in the system. As the separation went on, the intensity of the red band slowly went down. The second and third fractions had the strongest bands of mitragynine (Figure 7). This suggests that MIP had the ability to hold onto impurities for longer periods of time while selectively releasing mitragynine. The results of both experiments show that the MIP sorbent’s ability to separate things depends on a number of things. The ratio between the stationary phase and the extract is one important factor that has a direct effect on how well the separation works. To obtain a good separation, the right amount of sorbent is required. Using a vacuum in the column system is another important factor. The second experiment showed that using a vacuum made things work better by cutting the separation time in half (8 hours) compared to the first experiment. The column system in the second experiment also showed more stability since the fraction collection went up to the 30th fraction without phase separation, which had happened in the first experiment.
Elution Profile of Fractions 1–10 in the First (Top) and Second (Bottom) Experiment.

Quantification of Mitragynine and Isolation Efficiency
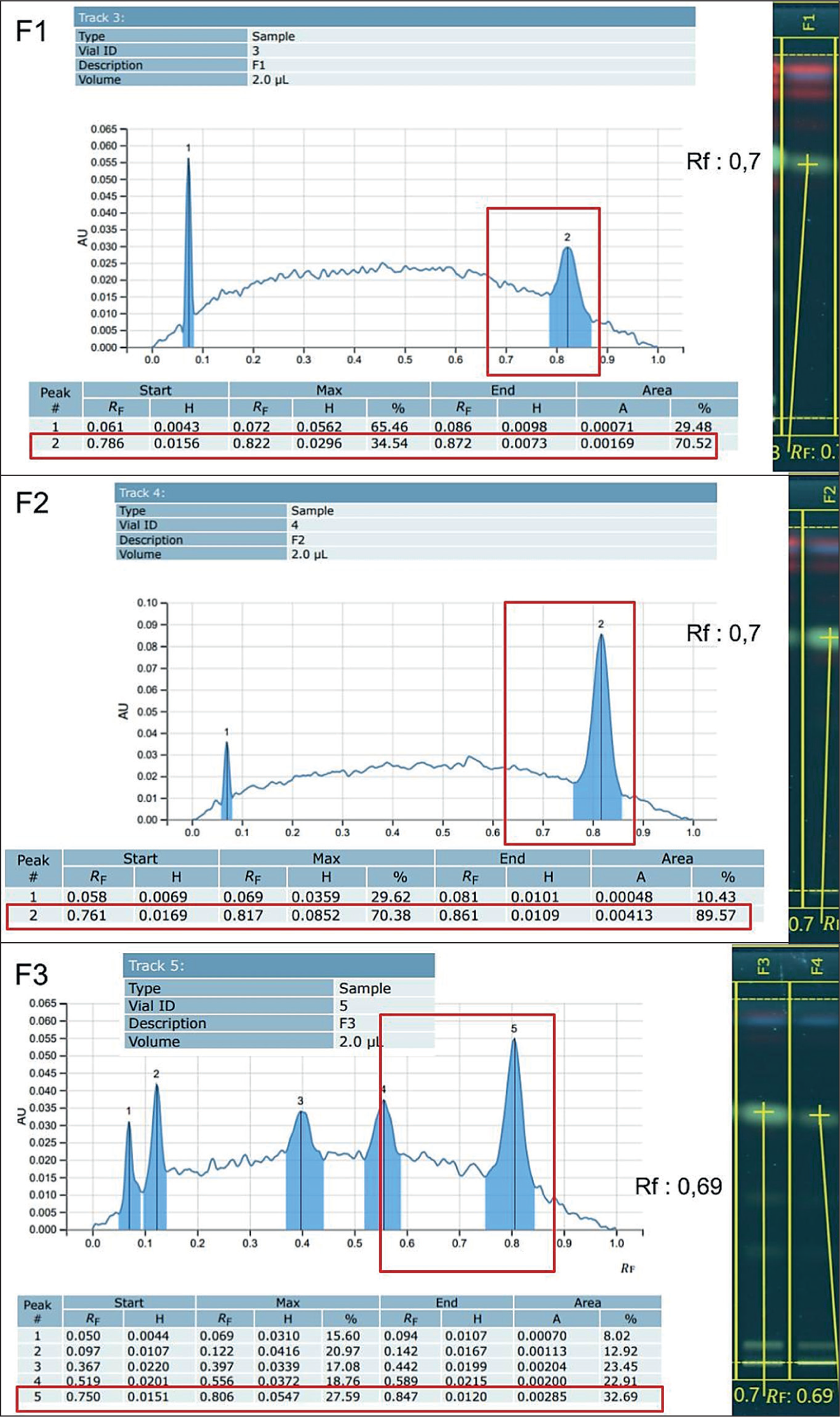
HPTLC analysis from the 10 fractions of the crude extract showed that only fractions 1 to 3 have bands that could be quantified by the system. This limitation was probably due to the small sample sizes and weak separation bands that were visible when characterized at, but not strong enough to be detected by the instrument. As a result, the method was not yet appropriate for the quantitative assessment of mitragynine, underscoring the necessity for additional instrument validation to determine the limit of quantification (LoQ) for mitragynine in TLC fractions. Since direct quantification was not possible, an alternative method was used that relied on predictive calculations. This process included comparing the AUC of the fractions compared to the AUC of mitragynine in the crude extract.
Then, it used published mitragynine concentrations to figure out how well the isolation worked. Dohárszky et al. (2024), Novianry et al. (2024), and Ernawati et al. (2024) found that the average concentration of mitragynine isolate in alcohol-based extracts is about 3.607%.36,37 Based on this, the theoretical yield of mitragynine from 250 mg of extract in this study is 9.018 mg. It is important to note that because the initial mitragynine concentration in the crude extract could not be directly quantified with the current HPTLC setup, this literature value was utilized strictly as a theoretical baseline. Consequently, the resulting isolation efficiency represents an estimated yield rather than an absolute measured recovery.
HPTLC analysis provided data for the crude extract and fractions 1–3 as shown in Figure 8. The AUC values for the crude extract, fraction 1, fraction 2, and fraction 3 at the Rf level for mitragynine were 0.02360, 0.00169, 0.00413, and 0.00285, respectively. Based on the previously reported literature concentration, the predicted amounts of mitragynine were 0.646 mg in fraction 1, 1.578 mg in fraction 2, and 1.089 mg in fraction 3. This calculation was performed by comparing the AUCs of fractions 1–3 to the AUC of the crude extract. The estimated total mitragynine recovered from the separation was 3.313 mg, which is 36.738% of the total amount of mitragynine in the crude extract that corresponds to the isolation efficiency. Additionally, a cost comparison was also performed to assess the cost of the method compared to the standard price of mitragynine from other vendors in this experiment. The standard price for mitragynine from the vendor is $200 per mg, with a purity of 92.94%. A preliminary cost estimate indicates that the raw materials required for synthesis, extraction, and isolation amount to approximately $58.28 per mg of crude isolate. However, it should be emphasized that this is strictly a material cost and does not represent a direct alternative to the commercial standard. This estimate excludes critical operational expenses, including equipment use, electricity, extensive downstream purification, and advanced characterization (e.g., MS and nuclear magnetic resonance [NMR]). While the MIP method demonstrates promising selectivity, further process optimization is necessary before a comprehensive cost-benefit analysis can be established.
Comparison Between the HPTLC Spectrum and the Separation Pattern of Fractions 1–3.
Isolate Characterization
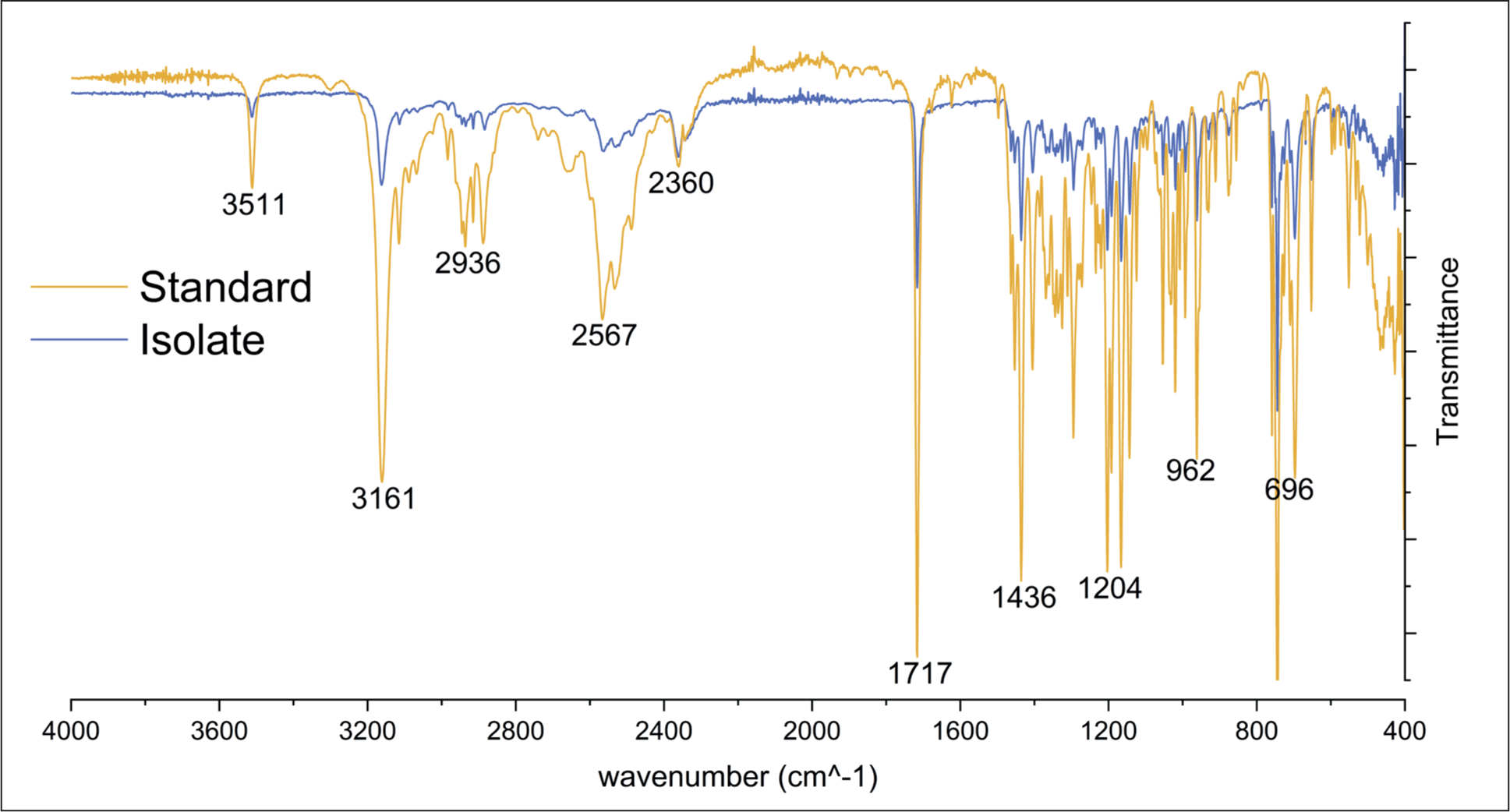
The mitragynine isolate was characterized using FTIR spectroscopy and compared to the standard mitragynine to confirm its structural identity. Both samples were analyzed using ATR, and spectra were recorded over a wavenumber range of 400–4,000 cm–1. The FTIR spectrum of the isolate exhibited characteristic peaks that were consistent with those observed in the mitragynine standard (Figure 9). Notable functional groups identified in both spectra included the N-H stretch at approximately 3,161 cm–1, the alkyl C-H stretch near 2,936 cm–1, the C=O carbonyl stretch at approximately 1,717 cm–1, and the C-N stretch within the range of 962 to 1,204 cm–1. Although the peaks in the standard sample exhibited higher intensity, this discrepancy is likely due to the presence of additional impurities or matrix components in the isolate, which may have resulted in a reduced response. Nonetheless, the corresponding peaks observed in both the standard and the isolate confirm the presence of mitragynine in the isolated sample. To quantitatively evaluate structural similarity, cosine similarity analysis was performed in R software by comparing the IR spectra of two samples. The analysis yielded a similarity score of 99.6%, indicating identical spectral correspondence between the isolate and standard mitragynine (Supplementary Materials 2). The FTIR analysis provides strong evidence supporting the successful isolation of mitragynine, highlighting that the MIP effectively binds to mitragynine, facilitating its selective isolation from the complex kratom matrix. These findings underscore the potential of MIPs as highly selective sorbents for extracting target compounds from complex natural product matrices, such as kratom, where mitragynine is present among many secondary metabolites. The ability of MIPs to selectively bind to mitragynine, even in the presence of other structurally similar compounds, represents a significant advancement in the extraction of alkaloids from complex plant matrices. This approach offers a potential alternative for isolating bioactive compounds like mitragynine, which is challenging to purify due to the similarities between kratom’s secondary metabolites. To further validate the purity and structural integrity of the isolated mitragynine, additional analytical techniques such as NMR and MS could be employed. These methods would provide complementary data, further confirming both the purity and structural identity of the isolated mitragynine, paving the way for more robust applications of MIP technology in natural product chemistry.\
Infrared Spectra of Isolated Mitragynine and the Standard.
Comparison with Other Methods
A comparison was made between the novel MIP method and conventional PTLC to highlight the advantages of MIPs, particularly in terms of selectivity and efficiency of isolation. Mobile phase optimization was conducted to determine which solvent would work best for the isolation process. Based on its capacity to distinguish the mitragynine spot from other compound spots, the ideal mobile phase was identified. A combination of mobile phases comprising n-hexane, ethyl acetate, and 25% ammonia was employed in this investigation. N-hexane has a polarity index of 0.1, making it a nonpolar solvent; meanwhile, 25% ammonia is a polar solvent with a polarity index of 7.72. Ethyl acetate, with a polarity index of 4.4, is semi-polar. A solution is considered more polar if its polarity index is higher. 38 Four mobile phase were optimized employing n-hexane:ethyl acetate:25% ammonia, with the following ratios: (30:15:1), (35:10:1), (37:8:1), and (25:20:1). The result indicated that mobile phase 1, 2, and 3 were able to elute or carry mitragynine, but only phase 1 effectively separated the mitragynine spot meanwhile mobile phase 4 did not effectively elute or carry mitragynine, as evidenced by mitragynine remaining at the starting point of the elution (Figure 10a). It can be concluded that the mobile phase 1 is the most optimal mobile phase for this separation. After the spot that contained mitragynine was scraped out and separated, a study was performed to ensure that the isolated compound was pure, without any impurities. The investigation was carried out using 2D TLC. To achieve optimal resolution, two different solvents were used. 39 The first mobile phase was n-hexane:ethyl acetate:25% ammonia (30:15:1), and the second mobile phase was n-hexane:ethyl acetate:25% ammonia (35:10:1). The results from both elutions showed only one spot with the same green color (Figure 10b). Therefore, it can be concluded that the isolate contains only mitragynine and no other compounds.
Mobile Phase Optimization of TLC (a) and 2D TLC Result (b).

The estimated total mitragynine recovered from the separation was calculated using the same previous MIP method, with 3.607% isolation efficiency. The low isolation efficiency can be attributed to several factors, including the limited sensitivity of HPTLC in detecting compounds at low concentrations. Only 7 out of 14 samples showed meaningful spots, which can be detected in the instrument within the Rf value of mitragynine in the extract when the analysis was performed at the peak area. This disparity might have happened as a result of the detector’s inability to detect some peaks or because the peaks that were detected were low, so they were categorized as noise or unimportant impurities. 40 In comparison, MIP-based separation showed enhanced selectivity and efficiency over PTLC, as evidenced by the faster separation process and higher purity of the mitragynine isolate. The MIPs method was able to selectively bind and isolate mitragynine from the complex matrix of kratom leaf extract, with reduced processing time and minimal sample loss. In contrast, PTLC required longer processing times and had lower selectivity due to the broad interaction of the silica gel with various compounds in the sample. These results demonstrate that MIPs offer significant advantages over traditional methods, providing a more efficient and selective approach for isolating mitragynine.
Study Limitations and Future Optimization Direction
The current study has several intrinsic limitations that should be acknowledged. One of the primary limitations is the use of colchicine as a dummy template molecule due to the limited availability of mitragynine. Although colchicine provided a viable alternative with sufficient structural similarity, future studies should aim to explore more suitable and highly specific dummy templates that more closely mimic mitragynine. This could lead to improved binding selectivity and higher efficiency in the MIP synthesis process. Additionally, the polymerization method and conditions, while optimized for this study, may still benefit from further refinement. The temperature, polymerization time, and initiator concentration all played crucial roles in determining the yield and quality of the MIP, but further studies should focus on fine-tuning these parameters to maximize the polymer’s stability, affinity, and reusability.
The isolation efficiency, although promising, was limited to 36.7%, suggesting that the method could be further optimized to increase the yield of mitragynine from kratom leaf extract. In particular, the extraction process, which utilized methanol as a solvent, could be evaluated against other solvents or solvent mixtures to determine whether higher recovery rates can be achieved. Moreover, a more extensive analysis of the separation conditions, including the flow rate during column chromatography and the application of vacuum assistance, could help to optimize the separation process and reduce the time required for isolation. Another limitation was the relatively small sample size in the isolation and quantitative analysis. Future studies should consider increasing the sample size and conducting parallel experiments to confirm the reproducibility and scalability of the MIP-based isolation technique. The use of more advanced analytical techniques, such as mass spectrometry or NMR, would provide a more detailed understanding of the isolate’s purity and structural identity, which would further validate the reliability and effectiveness of the MIP process. While this study demonstrates the potential of MIPs for selective mitragynine isolation, future optimization efforts should focus on improving the dummy template design, refining polymerization conditions, enhancing the extraction and separation processes, and validating the isolate’s structure using advanced analytical tools. These advancements could significantly enhance the efficiency, selectivity, and scalability of the method for isolating mitragynine complex kratom matrices.
Conclusions
This research presents a novel approach to mitragynine isolation using MIP, providing a more selective and efficient method compared to traditional techniques. The ability of MIPs to selectively bind to mitragynine, even in the presence of other structurally similar compounds, represents a significant advancement in the extraction of alkaloids from complex plant matrices. This approach offers a potential alternative for isolating bioactive compounds like mitragynine, which is challenging to purify due to the similarities between kratom’s secondary metabolites. The synthesis was optimized using a bulk polymerization process with a template-monomer-CL ratio of 1:3:20 at 80°C for 24 hours. The template molecule, colchicine, was efficiently removed via sonication, with the 16th cycle of methanol treatment, as confirmed by infrared spectroscopy. Adsorption experiments revealed that MIP conformed to a pseudo-second-order kinetic model and exhibited adsorption behavior consistent with the Langmuir isotherm, indicating a high degree of selectivity and stability in the binding sites. The effective isolation of mitragynine was validated through HPTLC, with a recovery of 3.313 mg of mitragynine, demonstrating an isolation efficiency of 36.7%. Infrared spectroscopy confirmed the structural identity of the isolate, showing a high degree of similarity (99.6%) to the standard mitragynine, further corroborating the successful isolation process. Additionally, the separation process utilizing MIP exhibited superior selectivity and efficiency when compared to traditional PTLC, which showed less precision in isolating mitragynine from the complex kratom matrix. These results indicate that MIPs are highly effective as selective sorbents for isolating mitragynine from plant extracts, offering an alternative to conventional separation methods that often lack specificity and efficiency. The use of MIP not only improved the isolation efficiency but also reduced the processing time compared to PTLC, suggesting its potential for large-scale application in the purification of alkaloids from plant sources. For future research, exploring more sophisticated dummy template molecules could further enhance the specificity and binding affinity of the MIP, leading to higher selectivity and improved recovery rates. Moreover, the integration of additional characterization techniques, such as NMR and mass spectrometry, is recommended to provide further validation of the isolate’s purity and structural integrity. These advancements could contribute to the development of more efficient and cost-effective methods for isolating mitragynine and other valuable alkaloids from complex plant matrices, with potential applications in pharmaceuticals and natural product chemistry.
Supplemental Material
Supplemental material for this article is available online.
Supplemental Material
Supplemental material for this article is available online.
Footnotes
Authors’ Contribution
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Data Availability Statement
All the data is available with the authors and shall be provided upon request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Approvals
This study does not involve experiments on animals or human subjects.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Directorate of Research and Community Services, Directorate General of Research and Development, Ministry of Higher Education, Science, and Technology of the Republic of Indonesia (Contracts No. 124/C3/DT.05.00/PL/2025; and No. 0982/LL3/AL.04/2025).
Informed Consent
Not applicable.
Use of Artificial Intelligence-assisted Technology
The authors used QuillBot, an AI tool, to help rephrase some parts of the manuscript and make it more readable. The tool was only used for improving the language; all the content and findings are based on the authors’ original research. The authors carefully reviewed and adjusted the AI-generated text to ensure its accuracy and clarity.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.