
Abstract
Autosomal recessive Limb-Girdle Muscular Dystrophy type R28 (LGMDR28; OMIM #620375) is one of the most recently identified subtypes of recessive LGMD. To date, 17 affected individuals from eight unrelated families have been reported to harbor biallelic variants in the HMGCR gene. Here, we report eleven individuals from six unrelated Middle Eastern families diagnosed with LGMDR28 and carrying homozygous variants in HMGCR. Our cohort recapitulated most of the previously described clinical features of the HMGCR-related phenotype. In addition, we also report a family with congenital onset of the condition as well as two affected individuals with liver failure. Through genome and exome sequencing, we identified three novel homozygous missense variants; c.2519G >A, p.(Arg840Gln), c.1784G >A, p.(Arg595His), and c.1783C>T, p.(Arg595Cys) and a homozygous in-frame deletion (c.1522_1524del, p.(Ser508del)) previously reported in compound heterozygous state with a second relevant variant and very recently as homozygous in an unrelated proband. This study consolidates the pathogenic role of HMGCR in LGMDR28 while expanding the phenotypic and mutations spectrum of LGMDR28.
Introduction
Limb Girdle Muscular Dystrophy (LGMD) is a sub-group of rare muscular dystrophies, collectively affecting approximately 1 in 61,000 individuals worldwide. The 39 subtypes identified to date exhibit significant genetic heterogeneity. 1 LGMD can be inherited following both dominant and recessive patterns, the latter being more common. The cardinal clinical feature of LGMD is progressive muscle weakness, primarily affecting the hip and shoulder muscles. Accurate diagnosis often requires a comprehensive evaluation by a neuromuscular specialist, including laboratory tests such as serum creatine kinase (CPK) measurements, genetic testing and, if need be, muscle biopsy. While genetic testing can confirm the diagnosis in around 30–70% of cases, 2 many individuals with LGMD remain genetically undiagnosed, suggesting the existence of additional causative genes.
Autosomal recessive LGMD type R28 (LGMDR28) is one of the most recently identified subtypes of recessive LGMD (OMIM# 620375). Two studies in 2023 identified the subtype in a total of six unrelated families, demonstrating variable age of onset and rate of disease progression.3,4 Very recently, Gunasekaran et al. (2025) reported two additional unrelated families with phenotypes consistent with previous cases. 5 All reported patients to date shared clinical features consistent with other forms of muscular dystrophy, including incidentally elevated serum CPK levels (>1000 U/L), proximal muscle weakness, variable age of onset, and disease progression leading to impaired ambulation. All published cases harbored biallelic variants in HMGCR, the gene encoding the HMG-CoA Reductase (HMGCR) protein (an enzyme) involved in the sterol-biosynthesis pathway (OMIM# 142910). The HMGCR enzyme converts HMG-CoA to mevalonate, and its activity is regulated by a negative feedback mechanism involving cholesterol and other end products of the related pathway. Cholesterol-lowering drugs, such as statins, work by inhibiting the HMGCR enzyme, in turn stimulating the hepatic synthesis of LDL receptors and lowering of LDL-cholesterol. In fact, SNPs in HMGCR have been associated with regulation of cholesterol/lipoprotein levels and response to statins.6,7
Here, we report six unrelated families from the Middle East: Two from Lebanon, two from Iran, two from Turkey, all with homozygous HMGCR variants. The families we report present with three novel missense homozygous variants and a homozygous in frame deletion which was reported previously in HMGCR, further strengthening the involvement of this gene in LGMDR28 and broadening the phenotypic spectrum of this condition.
Materials and methods
Patient recruitment
Six unrelated families presenting with a variety of clinical indications were aggregated from four different centres, Department of Human Genetics (Lebanese American University), Dr Shahrooei Lab (Iran), Department of Medical Genetics (Istanbul University) and Izmir Biomedicine and Genome Center (Dokuz Eylul University), by direct communication or via GeneMatcher. 8 Biological samples were collected after obtaining informed consent from patients or their parents. Additionally, informed consent was obtained from the parents or legal guardians of the patients/participants for the publication of their data.
Genome/exome sequencing
To select the disease-causing genetic variants in our probands, we performed either genome or exome sequencing based on the description below. Genetic variants are described following the Human Genome Variation Society (HGVS) recommendations.
9
The selected variants were classified according to ACMG
10
and ClinGen guidelines (https://clinicalgenome.org/working-groups/sequence-variant-interpretation/
Families 1 and 2: DNA was extracted from Buccal swabs and TruSeq NanoDNA High Throughput Library Prep Kit (Illumina®) was used to prepare libraries, which were sequenced using the 150nt paired-end protocol on an Illumina platform to yield an average coverage depth of 30× for the nuclear genome and minimum 1000× for the mitochondrial genome. Raw read alignment to reference genome GRCh38 and variant calling, including single nucleotide substitutions (SNVs), small insertions/deletions (Indels) and structural variants (SVs) with default parameters were performed using DRAGEN (version 4.2.4, Illumina). SNV and Indel variant annotation was performed by Geneyx (https://geneyx.com). Structural variants were annotated with ANNOTSV3.1 and an inhouse structural variant database to obtain allele frequencies.
Families 3–6: DNA was extracted from peripheral blood and exome sequencing was performed by Macrogen, South Korea. Libraries were prepared using Agilent SureSelect V7, and sequenced on the NovaSeqX Plus with a 150nt paired-end configuration, resulting in an average coverage depth of 100x. Raw reads were aligned to reference genome GRCh37 using BWA, and variants were called using the GATK package and annotated with ANNOVAR. Variants were filtered according to GATK's best practices and further filtered based on our in-house database and for variants with MAF <1%.
Sanger analysis
The relevant region of HMGCR based on GRCh38 reference sequence was amplified via the PCR method and directly sequenced using flanking or internal primer pairs. FinchTV was used for viewing and analyzing trace data from Sanger DNA Sequencing.
Results
Clinical assessment
A clinical summary of the affected members from the six families is presented in Table 1.
Demographics and clinical features of subjects reported with LGMDR28 and holding HMGCR variants.
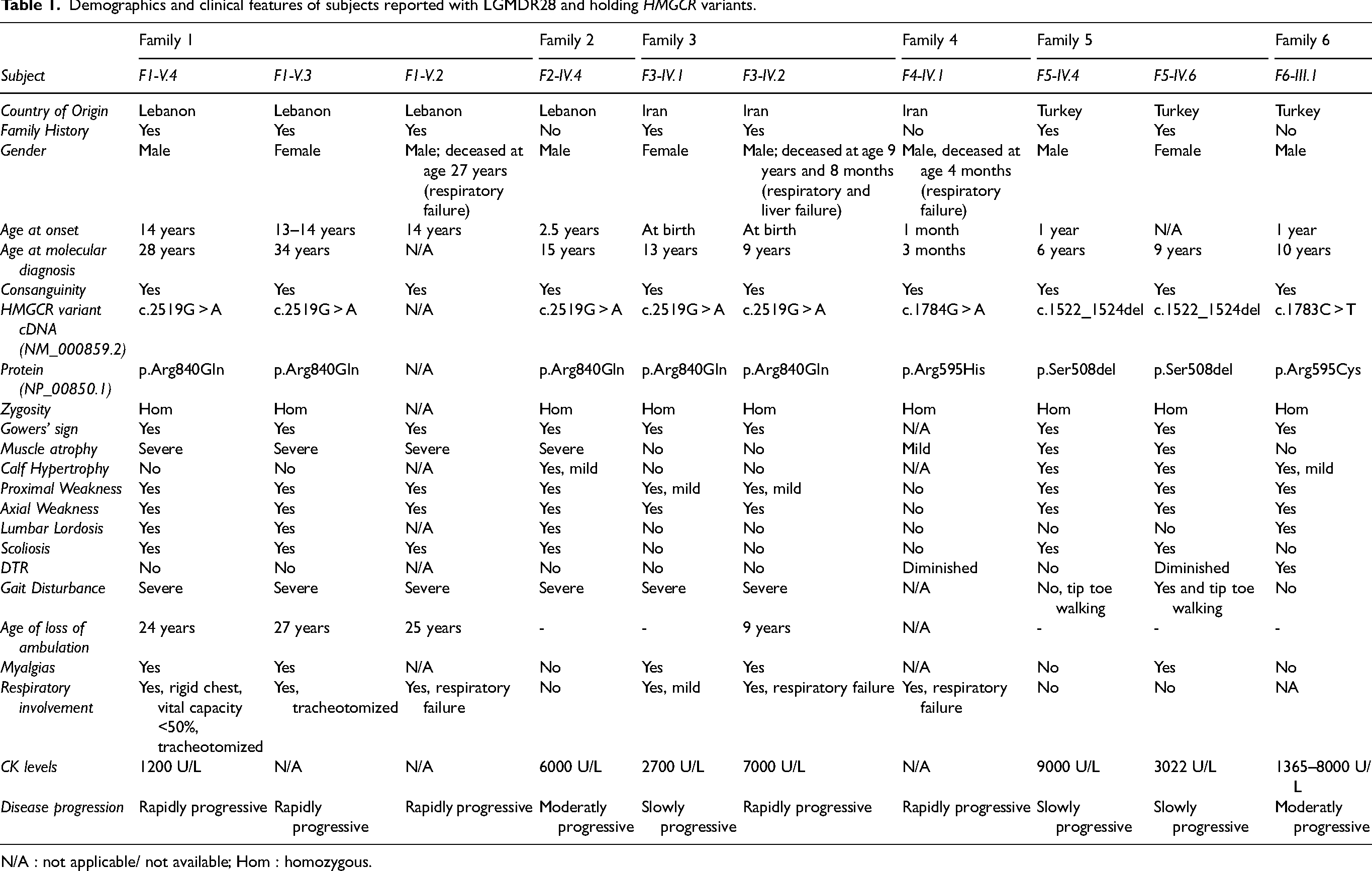
N/A : not applicable/ not available; Hom : homozygous.
Disease progression was classified as rapidly progressive when marked functional deterioration occurred within months to a few years from symptom onset, moderately progressive when a steady but noticeable decline occurred over several years, slowly progressive when minimal functional decline was observed over an extended follow-up period, and stable when no significant motor function or respiratory deterioration was documented over time.
For families 1, 2, 3, 4, and 6 gestation and delivery were unremarkable, with no history of exposure to pre- or perinatal environmental toxins, including statins. Birth weight, length, and occipitofrontal circumference (OFC) were within normal limits.
Family 1
Family 1 is a consanguineous Lebanese family (Figure 1A). For all three affected siblings, neonatal courses were uncomplicated, and developmental milestones as well as cognitive functions were reported as normal. Initial symptoms, including proximal and axial muscle weakness and fatigue, appeared between the ages of 13 and 14 years. Thoracolumbar scoliosis was noted between ages 15 and 16 years. All patients experienced rapid disease progression, leading to severe muscular atrophy, significant mobility decline, and respiratory difficulties necessitating ventilatory support and, eventually, tracheostomy. None of the three affected siblings exhibited involvement of other organs, particularly no signs of cardiomyopathy. A comprehensive clinical examination was available only for the proband; phenotypic information for the two other affected siblings was obtained retrospectively from family reports and medical records, as one sibling was bedridden and the other had died at 27 years of age due to respiratory failure and cardiac arrest (Table 1).
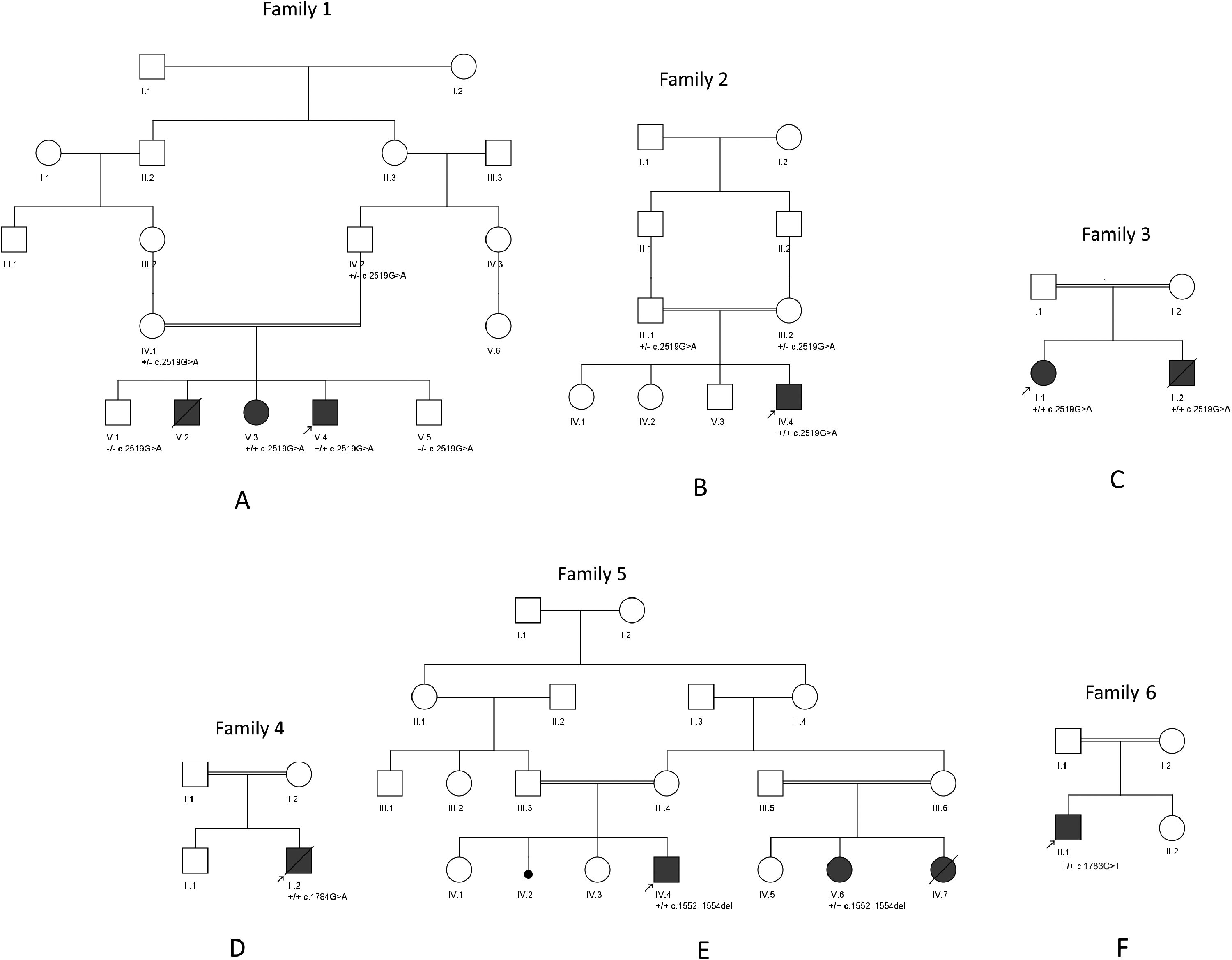
Pedigrees of the six families reported here.
We first saw the proband (Figure 1A, V.4) at age 23 years. He was wheelchair-dependent but could still take a few steps with support. He exhibited severe amyotrophy. His height was 175 cm, and his weight was 51 kg (BMI 16.7). Proximal and axial muscle weakness was evident, with absent deep tendon reflexes. Calf hypertrophy was not observed, and there were no signs of fasciculations nor macroglossia. Ophthalmological examination was normal. Serum CPK level was 1200 U/L. Muscle biopsy was not performed. Based on the initial clinical suspicion of limb-girdle muscular dystrophy, we conducted sequence analysis and deletion/duplication testing of the 117 genes present in the Neuromuscular Disorders Panel which turned out to be inconclusive.
The patient was reevaluated at age 25 years. He reported completely losing ambulation a year prior.
He presented with multiple joint contractures and muscle wasting. Deep tendon reflexes remained absent. The chest was rigid, and forced vital capacity was estimated at less than 50% of the predicted value, thus requiring nocturnal invasive ventilation. A subsequent exome sequencing was performed and the results were negative. At a follow-up at age 29 years, the proband could only move his fingers and exhibited further muscle atrophy. He was bedridden, severely underweight, and fully dependent on a ventilator.
Family 2
The proband is the fourth child of first cousin Lebanese parents (Figure 1B, IV.4). The initial development was uneventful and motor milestones were achieved in due course. Soon after achieving independent ambulation at around 13 months, parents noticed muscular weakness and some fatigue with the child getting tired very quickly. The clinical examination was rather inconclusive at that stage. The proband exhibited slightly enlarged calf muscles, mild hyperlaxity of the ankles and knees, and an incomplete Gowers’ sign, characterized by the use of the hands for partial support when rising from the floor, without the full “climbing up the thighs” maneuver. Serum CPK levels were measured around 4000 IU/l on various occasions. A muscular biopsy was performed but no definite conclusions could be drawn from it. The main immunostainings (against dystrophin, sarcoglycans and dysferlin) were reported as normal. At age 2.5 years, muscle weakness was still present with, additionally, a noticeable weakness of neck flexors. The Gowers’ sign was fully positive. Brain MRI was reported as normal.
We first examined him at age 8 years, when he presented with fatigue and difficulty climbing stairs or performing physical activities like his peers. His height was 129 cm (60th percentile), and his weight was 20 kg (3rd percentile) (BMI: 12). He displayed global muscular hypotrophy, neck muscle weakness, proximal and axial muscle weakness, mildly tense calves, and absent deep tendon reflexes. Gowers’ sign was positive. Ophthalmological examination was normal. Laboratory tests revealed an elevated CPK level of 6000 IU/L. Sequence analysis and deletion/duplication testing of the 117 genes present in the Neuromuscular Disorders Panel designed were performed and were negative.
At age 15 years, he presented with a marfanoid-like global amyotrophy, and a long neck. His gait was unsteady and slow with significant lumbar lordosis. Muscle weakness was more pronounced, and he could no longer stand from a chair without assistance. Deep tendon reflexes were absent. Mild calf hypertrophy was observed. A skeletal survey showed a 47° lumbar scoliosis associated with significant coronal imbalance (lateral trunk shift) and sagittal imbalance (abnormal sagittal vertical axis).
Family 3
The proband is the first child born to a consanguineous Iranian couple (Figure 1C, IV.1). At birth, she showed limb myopathy. At age 3 years, muscle biopsy findings showed myopathic atrophy with multiple dispersed non-neurogenic pattern fibers, and EMG findings revealed a myopathic process involving mostly proximal muscles of lower limbs. At age 10 years, she started exhibiting shortness of breath. Accordingly, and because of symptoms related to a myopathic process and the involvement of the liver, she was suspected to be affected by mitochondrial myopathy. When examined at 13 years, she displayed normal intelligence but showed axial weakness, mild proximal muscle weakness, absent deep tendon reflexes, positive Gowers’ sign, toe walking, mild respiratory difficulties and serum CPK levels were measured at around 2700 U/L. Through occupational therapy, her walking and gait have shown improvement over time. She also suffers from progressive liver cirrhosis, which has necessitated consideration for a liver transplant. No history of liver problems has been reported in the pedigree.
The proband's younger brother (Figure 1C, IV.2), was seen at the age of 9 years and 6 months. He similarly presented with muscle weakness since birth and was suspected to be affected by mitochondrial myopathy. He had positive Gowers’ sign, severe gait disturbance and had lost ambulation at the age of 9 years. In addition, serum CPK levels were measured at around 7000 U/L. Following a minor medical incident, he was diagnosed with thrombocytopenia and progressive liver cirrhosis, which required transplantation. He also developed significant acute respiratory distress and regression in both his verbal and motor skills. He was hospitalized due to these complications, required ventilatory support, and passed away due to respiratory arrest and heart failure at the age of 9 years and 8 months.
Family 4
The proband is a male infant born to a consanguineous Iranian couple (Figure 1D, IV.2). At 1 month of age, he exhibited diminished deep tendon reflexes and mild muscle atrophy. He was admitted to the hospital due to significant hypotonia, severe congenital myopathy, and respiratory distress, which necessitated his reliance on a ventilator for adequate breathing support. Since then, he has remained dependent on mechanical ventilation. The patient had an older brother who is healthy. The patient passed away at the age of 4 months. There are no other known hereditary disorders present in his family history.
Family 5
Two first-degree cousins from a consanguineous Turkish family were referred to the clinic with proximal muscle weakness and suspicion of myopathy. The proband (Figure 1E, IV.4) was a 6-year-old male who showed delayed motor milestones, hypotonia, and decreased muscle strength. He had a history of sucking difficulties. Although motor development was delayed, he showed normal speech development. Serum CPK level was found to be elevated up to 9000 IU/l.
His affected cousin (Figure 1E, IV.6) was born at 33 weeks of gestation following oligohydramnios. She had normal motor and verbal milestones. However, CPK was found to be elevated, with progressively rising levels. At 9 years of age, she was diagnosed with the same symptoms as her cousin, including Gowers’ sign, calf hypertrophy, scoliosis, and proximal and axial weakness. The progress of disease was slow in both, proband and affected cousin, up until the last examination in December 2024. The younger sister of the affected cousin (Figure 1E, IV.7) had a more severe course and died from respiratory failure. A glycogen storage disease type 4 was suspected after a liver biopsy revealed possible glycogen storage. However, there remains uncertainty as to whether she was affected by LGMDR28 like her sister (Figure 1E, IV.6), by a glycogen storage disease, or by both conditions simultaneously.
Family 6
The proband is currently 10 years old and has a history of a normal, term birth to consanguineous parents. At 21 months of age, the patient was initially evaluated due to an incidental finding of elevated CPK levels. The patient's medical history revealed delayed motor developmental milestones. There was no known family history of neuromuscular diseases. The proband, which was followed up with axial hypotonia, motor delay, and elevated CPK levels, with values fluctuating between 1365 and 8000 U/L. Investigation for mutations in the DMD gene using MLPA and Sanger sequencing yielded negative results. The LGMD panel was also negative. Metabolic tests were normal. Nerve conduction studies (NCS) were normal, and needle electromyography (EMG) revealed myopathic changes in the proximal muscles. At the age of 6 years, the patient exhibited muscle strength of 4/5 in the neck flexors and extensors, as well as in the proximal lower extremities. A muscle biopsy performed at the age of 7 years showed myopathic changes, with no abnormalities detected on immunohistochemical examination, no dystrophic changes, normal collagen VI staining, and McArdle disease excluded. Muscle MRI of the lower extremities at 9 years of age shows mild, asymmetric STIR signal increase in selected thigh and calf muscles, slightly more pronounced on the right side. These findings are suggestive of very early or low-grade myopathic involvement but remain nonspecific (Supplementary Fig. 1). On a follow-up examination during the same period, muscle strength in the neck flexors and extensors was 4/5, while muscle strength in the proximal lower extremities was 3/5. Lumbar lordosis was increased, and mild calf pseudohypertrophy was observed. Spirometry at the age of 10 years demonstrated a mildly reduced FVC (1.78 L, 80% predicted, Z = –3.04) with only mildly decreased FEV1 (1.65 L, 89% predicted, Z = –1.52). The FEV1/FVC ratio remains elevated (92%, 109% predicted, Z = + 1.85), a pattern suggestive of a restrictive ventilatory defect, though submaximal inspiratory effort cannot be excluded. The patient had a mildly progressive course with a gradual decline over years.
Genetic analysis
DNA from probands of families 1 and 2 were subjected to genome sequencing, while those from probands of families 3, 4, 5 and 6 were subjected to exome sequencing. Variants in the HMGCR gene (NM_000859.2) were identified in all six families. Details of variant annotations are available in Table 2. In three unrelated families (1–3), the probands were found to carry a novel homozygous missense variant, c.2519G>A p.(Arg840Gln). This variant, located in exon 19, has a minor allele frequency (MAF) of 0.0000119 in gnomAD (10 carriers and no homozygotes). The CADD 11 and REVEL 12 scores for this variant were 23.4 and 0.14, respectively. Sanger sequencing confirmed the presence of this variant in a homozygous state in the affected siblings and a heterozygous state in the parents; patient IV.7 from family 5 could not be tested for the variant. The proband in family 4 presented with a novel homozygous missense variant, c.1784G > A, p.(Arg595His) located in exon 14 with a MAF in gnomAD of 0.000004956 (8 carriers and no homozygotes). The CADD and REVEL scores w ere 29.7 and 0.597, respectively. In the fifth family, both affected individuals presented with an in-frame deletion, c.1522_1524del, p.(Ser508del), located in exon 12. This variant has been reported previously as causal in a family affected with LGMDR28. 3 The proband in family 6 showed a novel homozygous missense variant, c.1783C > T, p.(Arg595Cys) located in exon 14 with a MAF in gnomAD of 0.000008054 (13 carriers and no homozygotes). The CADD and REVEL scores were 33 and 0.430, respectively.
Annotation of HMGCR variants identified in this study.

*No homozygotes reported.
Ht, Heterozygous; H, Homozygous; VUS, Variant of Uncertain Significance; MAF, Minor Allele Frequency.
Discussion
The HMGCR gene encodes the 3-hydroxy-3-methylglutaryl-CoA reductase, a key enzyme involved in the sterol-biosynthetic pathway. Cholesterol-lowering drugs, such as statins, work by inhibiting the HMGCR enzyme, in turn stimulating the hepatic synthesis of LDL receptors and lowering of LDL-cholesterol. Continuous use of statins has a well-documented association with the development of myotoxicity and myopathy in a significant number of patients. Although the specific mechanisms of Statin Associated Muscle Symptoms (SAMS) remains unclear, there is increasing evidence for the role of the HMGCR enzyme, and its downstream metabolite, mevalonate, in maintaining muscular structure and/or function. 13 The presence of anti-HMGCR enzyme antibodies has been found to result in auto-immune necrotizing myopathy, a disease mimicking muscular dystrophy in adults but also in a growing number of children.14–16 This, along with the recent reports of HMGCR variants causing a distinct form of LGMD supports a significant role for the HMGCR enzyme in muscle function. In fact, recent knockdown studies in animal models have confirmed the critical role of HMGCR in skeletal muscle development and demonstrated that overexpression of pathogenic HMGCR variants fails to rescue the phenotype. 5 To date in the literature, six patients from a large consanguineous Bedouin family, 4 and eleven patients from seven unrelated families,3,5 all carrying biallelic mutations in HMGCR have been reported. Furthermore, Yogev and colleagues have shown that oral administration of synthesized and purified mevalonolactone for an affected individual resulted in symptoms improvement. 4 Our study introduces six more unrelated families with three novel missense variants to support the pathogenic role of HMGCR in LGMDR28.
The ultra-rare c.2519G>A, p.(Arg840Gln) genetic variant was identified in three of the current study's families. Protein stability studies using iMutant, a protein stability predicting tool, also show reduced stability for the variant protein. 17 Arg840, located within the catalytic section of the HMGCR enzyme, lies within the Lα10 structural element, a 27-residue α-helix that forms the core of the L-domain fold (Figure 2A). This distinct structure is unique to the HMGCR enzyme and helps define the overall shape and stability of the domain, as well as modulate its binding affinity with substrates. 18 The catalytic portion of the HMGCR enzyme is essential for carrying out the conversion of HMG-CoA to mevalonic acid. Although this missense variant is not predicted to be disease-causing by in silico tools, it shows segregation in three unrelated families with shared clinical features. The occurrence of this variant in these families is interesting. The two Lebanese families originate from different regions of the country, but both are from the Shiite community, as is the Iranian family. Since there are individuals of Arab ancestry in Persia, this may suggest a common ancestor.
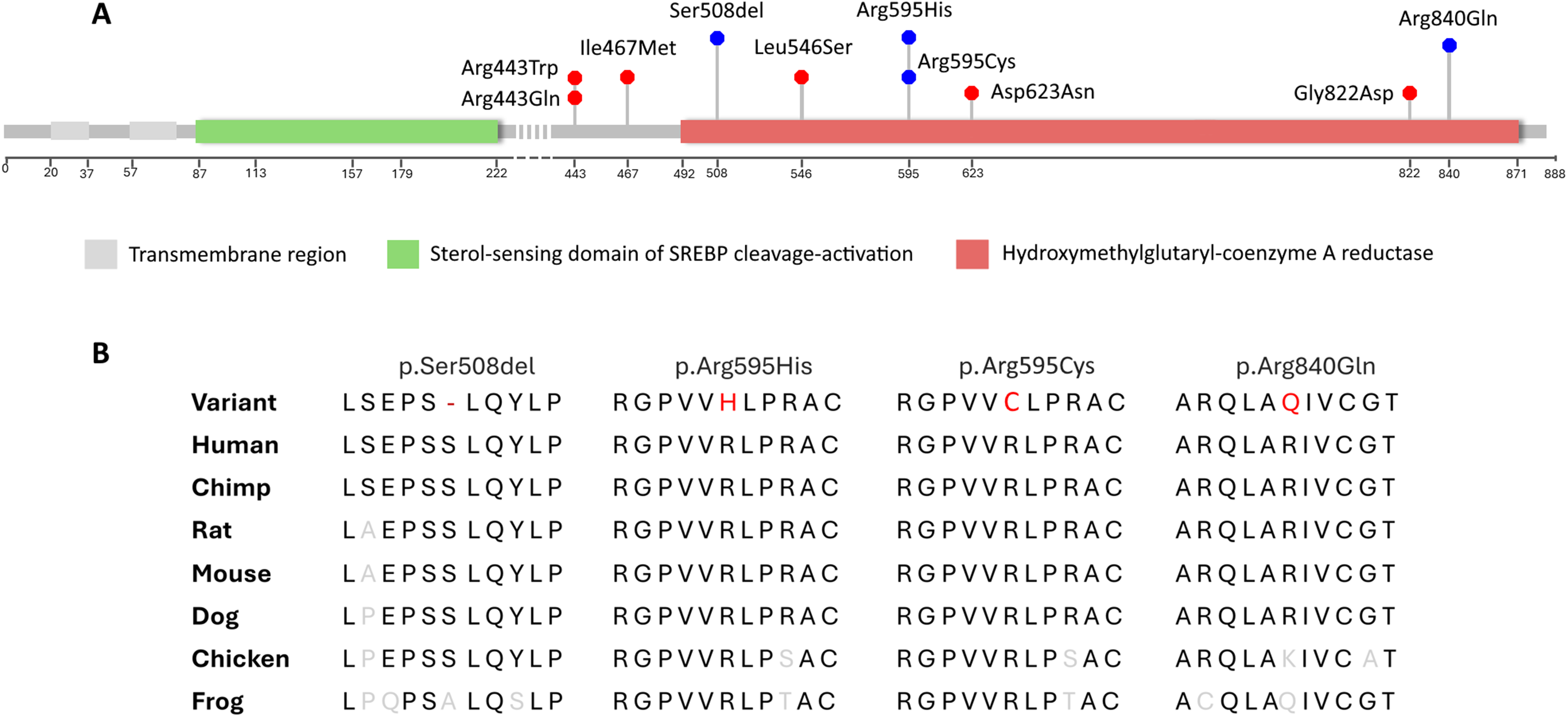
A- Lollipop chart showing the positions of all reported causal variations in HMGCR in individuals with LGMDR28. Blue circles represent variants reported in this study; B- Cross-species comparison of HMGCR sequences, showing the conserved nature of the variant residues.
The genetic variant identified in the proband of families 4 and 6, p.Arg595His, and p.Arg595Cys, are ultra-rare variants and both are predicted to be deleterious by in silico tools such as SIFT, Polyphen and CADD (Phred-scaled score: 29.7, and 33.0, respectively). The Arg595 residue is very well conserved across species and is also located in the catalytic domain of the enzyme (Figure 2). The proband in family 4 had a severe form of the disease with an early onset at 1 month, followed by a rapid clinical deterioration showing a pronounced progression in muscle weakness and atrophy, culminating in respiratory failure and death at 4 months. The proband in family 6 also had an early onset of phenotypes. In comparison with other reported patients in previous studies (Supplementary Table 1) and other patients in this study, both probands exhibited congenital/infancy onset, confirming the wide range of onset from birth to the fourth decade.
As with other LGMD subtypes, 1 we note in this new cohort a wide variability of clinical features. Scoliosis and/or lumbar lordosis were seen in families 1, 2, 5, and 6 and have not been reported as features in previously reported LGMDR28 patients. Deep tendon reflexes were reduced in all reported families except family 6. None of the patients had any cardiac involvement, as was the case in previous reports. It is important to note that early (even as early as neonatal) onset does not seem to correlate with severity of progression based on our cohort and this agrees with previous reports. 3 In fact, we see a marked range of disease severity and progression among patients with the c.2519G>A variant. The age of onset ranges from birth in the case of family 3 to as late as 14 years in family 1. We find both inter- and intra-familial variability is visible in the severity of respiratory involvement and in disease progression. In addition, both patients in Family 3 demonstrated bona fide hepatic involvement, a feature not observed in the other patients. Importantly, previously reported patients with HMGCR-related myopathy and other LGMDs have frequently shown elevated serum transaminase levels in the context of markedly elevated CPK, reflecting muscle injury rather than primary liver disease. Therefore, for individuals diagnosed with LGMDR28 we recommend monitoring not only CPK but also a broader liver panel (bilirubin, GGT, alkaline phosphatase, albumin, and INR), with further hepatology evaluation and imaging as indicated.4,19,20
This report also highlights the importance of periodically revisiting molecular results and unsolved diagnoses. The c.2519G>A variant that we describe here is not predicted strongly to be deleterious by most in silico tools and was overlooked in the analysis of NGS data. The utility of re-analyzing non-elucidated cases considering new data coming from next generation sequencing studies has repeatedly been emphasized.21–23
Based on reported patients (Supplementary Table 1), the gene-disease relationship is categorized as moderate by the ClinGen expert panel. 24 To date, all reported variants are missense or in-frame, except for a splicing variant whose consequences are not clarified. The current study provides additional evidence for the pathogenic role of this gene in LGMD. However, the mechanism of the disease remains unknown. Osaki and colleagues demonstrated that oral mevalonate supplementation rescued the myopathic phenotype in mice. 25 Additionally, the clinical study by Yogev and colleagues showed that oral administration of mevalonolactone in one patient improved symptoms. 4 Further functional studies and cases series are required to understand the exact mechanism of disease for this gene.
In summary, this study strengthens the disease-causing role of HMGCR in LGMDR28, illustrates a wide range of symptom onset from birth to the fourth decade, confirms the absence of heart issues, and raises the possibility of liver involvement.
Supplemental Material
sj-xlsx-1-jnd-10.1177_22143602261436296 - Supplemental material for Expanded clinical and genetic characterization of autosomal recessive HMGCR-related muscular dystrophy
Supplemental material, sj-xlsx-1-jnd-10.1177_22143602261436296 for Expanded clinical and genetic characterization of autosomal recessive HMGCR-related muscular dystrophy by Stephany El-Hayek, Aboulfazl Rad, Sahar Sedighzadeh, Gozde Yesil, Sandra Sabbagh, Mohammad Shahrooei, Pratibha Nair, Asuman Gedikbaşı, Sami Bizzari, Murtadha Ali, Eliane Chouery, Cybel Mehawej, Ayca Aslanger, Pejman Rohani, Meisam Sharifzadeh, Sinan Akbas, Javad Mohammadi-asl, Mahdiyeh Behnam, Sandra Corbani, Volkan Karaman, Uluç Yiş, Yavuz Oktay, Ipek Polat, J Andoni Urtizberea, Henry Houlden, Reza Maroofian, Gabriela Oprea and Andre Megarbane in Journal of Neuromuscular Diseases
Supplemental Material
sj-pptx-2-jnd-10.1177_22143602261436296 - Supplemental material for Expanded clinical and genetic characterization of autosomal recessive HMGCR-related muscular dystrophy
Supplemental material, sj-pptx-2-jnd-10.1177_22143602261436296 for Expanded clinical and genetic characterization of autosomal recessive HMGCR-related muscular dystrophy by Stephany El-Hayek, Aboulfazl Rad, Sahar Sedighzadeh, Gozde Yesil, Sandra Sabbagh, Mohammad Shahrooei, Pratibha Nair, Asuman Gedikbaşı, Sami Bizzari, Murtadha Ali, Eliane Chouery, Cybel Mehawej, Ayca Aslanger, Pejman Rohani, Meisam Sharifzadeh, Sinan Akbas, Javad Mohammadi-asl, Mahdiyeh Behnam, Sandra Corbani, Volkan Karaman, Uluç Yiş, Yavuz Oktay, Ipek Polat, J Andoni Urtizberea, Henry Houlden, Reza Maroofian, Gabriela Oprea and Andre Megarbane in Journal of Neuromuscular Diseases
Footnotes
Acknowledgements
The authors would like to express their deepest gratitude to the families involved for their full cooperation throughout the study. IP received support from an MRC strategic award to establish an International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD) MR/S005021/1 and ICGNMD supported data generation for one family.
ORCID iDs
Ethics approval
This study was conducted according to the ethical principles established by the World Medical Association Declaration of Helsinki. Ethical approval for the study was granted by the Institutional Review Board at the Lebanese American University (IRB #: LAUMCRH.AM3.2023.R1.1/Feb/2024).
Consent to participate/consent for publication
Written informed consent was obtained from the parents of all patients for the publication of this report. Informed consent for genetic analysis was obtained from the families in compliance with national ethics regulation.
Author contributions
SEH, AR, PN, SB, AU, EC, CM, and AM conceived, designed the study, performed data interpretation, wrote and edited the manuscript.
SSe, GY, SSa, MS, AG, MA, AA, PR, MSh, SK, JM, MB, SC, VK, UY, YO, IP, HH, RM, GO performed data interpretation, and edited the manuscript.
AR, GO, EC, CM contributed to data analysis.
All authors have read and approved the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.