
Abstract
Background:
Administration of Nusinersen requires repeated lumbar intrathecal access, posing challenges for patients with Spinal Muscular Atrophy (SMA). A purpose-built system may streamline drug delivery.
Objective:
To assess the feasibility and safety of ThecaFlex DRx for intrathecal Nusinersen dosing.
Methods:
We present initial results of a prospective, multicenter investigational device exemption (IDE) study. Patients with SMA who had an indication for intrathecal Nusinersen were enrolled. Primary outcomes were successful system implantation and postoperative infusion. Prespecified safety outcomes consisted of adverse events adjudicated for severity and device or procedure relatedness.
Results:
Twenty-five subjects underwent device implantation. Median age at implantation was 13.8 years (IQR 10.0–18.0), and 52% were female. Subjects included individuals with SMA types I (16%), II (64%), and III (20%). Implantation was successful in all cases. At the interim data cutoff, median follow-up was 230 days (IQR 76.0–369.0) at which point 23 subjects (92%) had successfully received Nusinersen infusions. Ten patients reached one year of follow-up, and all of them maintained a functional device at this visit. Sixty adverse events occurred in 19 subjects, with 12 events (20%) adjudicated as serious adverse events that were most commonly wound-related or respiratory. At one year, the estimated probability of remaining free from device action was 86.2% (95% CI 0.73–1.00), with two devices requiring explantation due to wound dehiscence and access difficulties, respectively.
Conclusions:
This interim report supports the feasibility of ThecaFlex DRx for Nusinersen administration, with long-term durability and effectiveness to be defined with longer follow-up.
For more information about the PIERRE study (NCT05866419), visit: https://www.clinicaltrials.gov/study/NCT05866419
Keywords
Introduction
Spinal Muscular Atrophy (SMA) is caused by biallelic deletions or mutations in the Survival Motor Neuron 1 (SMN1) gene, resulting in deficient SMN protein production and progressive apoptosis of alpha motor neurons in the spinal cord and brainstem. 1 SMA is classified into four clinical types that vary in onset and severity. All share the hallmark of progressive atrophy of voluntary and involuntary muscle groups. 2 The reported incidence is estimated at 1 in 11,000 to 15,000 live births, and is more frequently observed among individuals of European ancestry.3,4
Over the past decade, the U.S. Food and Drug Administration (FDA) has approved several disease-modifying therapies for SMA, with Nusinersen, an antisense oligonucleotide that promotes production of full-length SMN protein, as one of the available therapeutic alternatives. In the pivotal ENDEAR trial, Nusinersen significantly improved survival and motor milestone acquisition in infants with SMA type I compared with controls. 5 Subsequent studies, including long-term and real-world cohorts, have demonstrated sustained motor improvement several years after initiation of treatment.6,7
Because Nusinersen does not efficiently cross the blood brain barrier, treatment is delivered by intrathecal infusion into cerebrospinal fluid. Standard dosing involves six administrations in the first year and three annually thereafter. In this context, intrathecal dosing can be technically demanding in individuals with scoliosis or a history of spinal fusion: altered vertebral alignment and spinal instrumentation can complicate a safe needle trajectory into the intrathecal space, increasing procedure time and the likelihood of multiple needle passes. When radiologic guidance is required, cumulative exposure to ionizing radiation may increase over the course of therapy. In addition, repeated sedation or general anesthesia may be needed, introducing anesthesia related risks over time. 8
Grounded in such clinical hurdles, clinicians have sought a simpler mode of intrathecal administration of Nusinersen. The goal has been to develop a device that would allow repeated access to the intrathecal space without compromising safety. 9 Multiple intrathecal delivery strategies that have been successfully utilized in other clinical contexts have been repurposed for Nusinersen administration. 10 However, available small-scale reports of these non-specific intrathecal devices have noted concerns regarding device malfunction and longevity.11,12 In this study, we present our initial experience with a proprietary implantable catheter-port system designed specifically for intrathecal Nusinersen administration in patients with SMA.
Methods
Objective and study design
In 2023, the Study of an Intrathecal Port and Catheter System for Subjects With Spinal Muscular Atrophy (referred to as the PIERRE study) was initiated. This multicenter, single arm, investigational device exemption (IDE) study began enrollment at four institutions, with an additional site activated subsequently. Participants included in the present report were enrolled across these five sites. The objective of the study is to assess the safety and performance of ThecaFlex DRx (Alcyone, Inc., Lowell, MA) for intrathecal administration of Nusinersen in patients living with SMA. We present the first planned performance review of the ThecaFlex DRx in patients recruited from November 2023 through April 2025. Reporting is consistent with the CONSORT extension for pilot and feasibility trials where applicable. 13
Population
Following institutional review board approval and activation at the first four participating sites (Children's Hospital of Orange County, Boston Children's Hospital, Children's Hospital of Philadelphia, and Texas Children's Hospital), eligible subjects and legal guardians provided informed consent and subsequently underwent screening. Subjects were eligible for enrollment if they had a confirmed diagnosis of SMA and were 3 years of age or older. Other inclusion criteria were candidacy for treatment with Nusinersen and resistance to lumbar puncture, or other factors suggesting benefit from an implantable catheter-port system.
Exclusion criteria were contraindications for the use of ThecaFlex DRx, history of intrathecal delivery devices in the last six months, pregnancy, or any condition that interfered with outcome assessment. Once screened, subjects needed to be physically able to tolerate the implantation procedure. Full criteria may be consulted at ClinicalTrials.gov (NCT05866419).
Device description
The ThecaFlex DRx is a catheter–port system specifically designed for repeated intrathecal delivery of Nusinersen. It comprises an implantable intrathecal catheter connected to a subcutaneous port via a locking interface and incorporates a dedicated fixation device (Figure 1). The thermoplastic polyurethane catheter is designed for durability and kink resistance. This section possesses 31 outlet holes arranged circumferentially in a spiral configuration, with an additional terminal tip hole intended to preserve egress when the catheter tip abuts tissue. Three radiopaque marker bands facilitate fluoroscopic visualization.
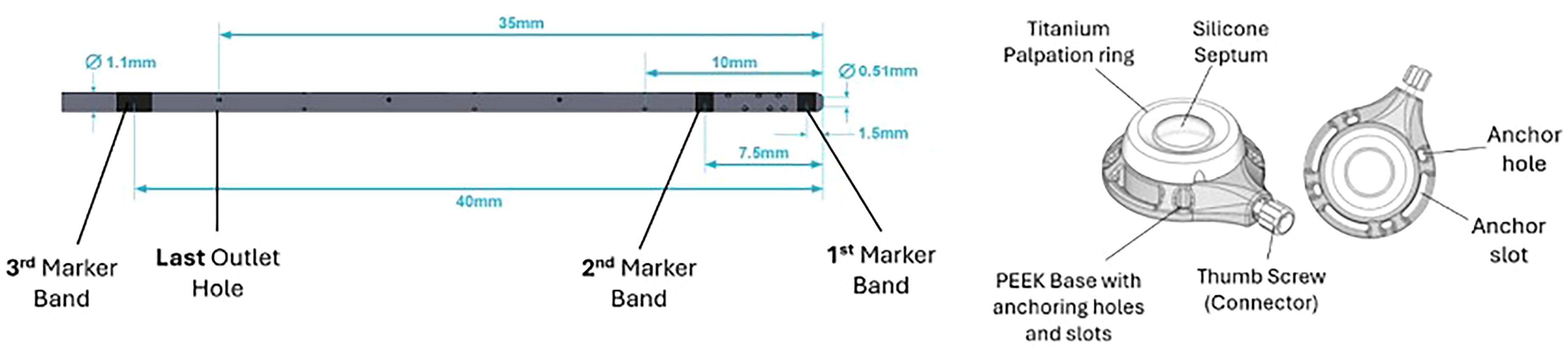
Device image. Architecture of the port and catheter system used in this study. Distal catheter segment with three radiopaque marker bands and a fenestrated tip. The implantable port is made of PEEK and has a silicone septum for injection. An anchor hole and an anchor slot provide options for fascial fixation. Drawing not to scale.
The subcutaneous port has a flat, low-dead-volume profile with a titanium-reinforced septum to facilitate access. The catheter–port connector is designed to reduce the risk of disconnection, and the dedicated fixation device anchors the assembly to help minimize migration. The multi-orifice distal segment is intended to maintain infusion delivery despite local tissue contact, addressing previously reported obstruction in rostrocaudal drug distribution with other catheters. 9
Surgical procedure
Surgical implantation technique parallels that of other intrathecal delivery systems, including the Ascenda catheter (Medtronic, Minneapolis, MN). 14 Intrathecal access is achieved by transfascial lumbar puncture using the needle provided in the system kit. The catheter is usually placed at mid to higher lumbar levels (Figure 2). To ensure appropriate intrathecal positioning, a minimum of 7 cm of catheter is advanced into the subarachnoid space. Fluoroscopy or plain radiography are used to confirm that the third marker band, located 5 mm proximal to the last outlet, is positioned at or below the L1 vertebral body. This ensures that the medication is delivered into the lumbar or thoracolumbar subarachnoid space.
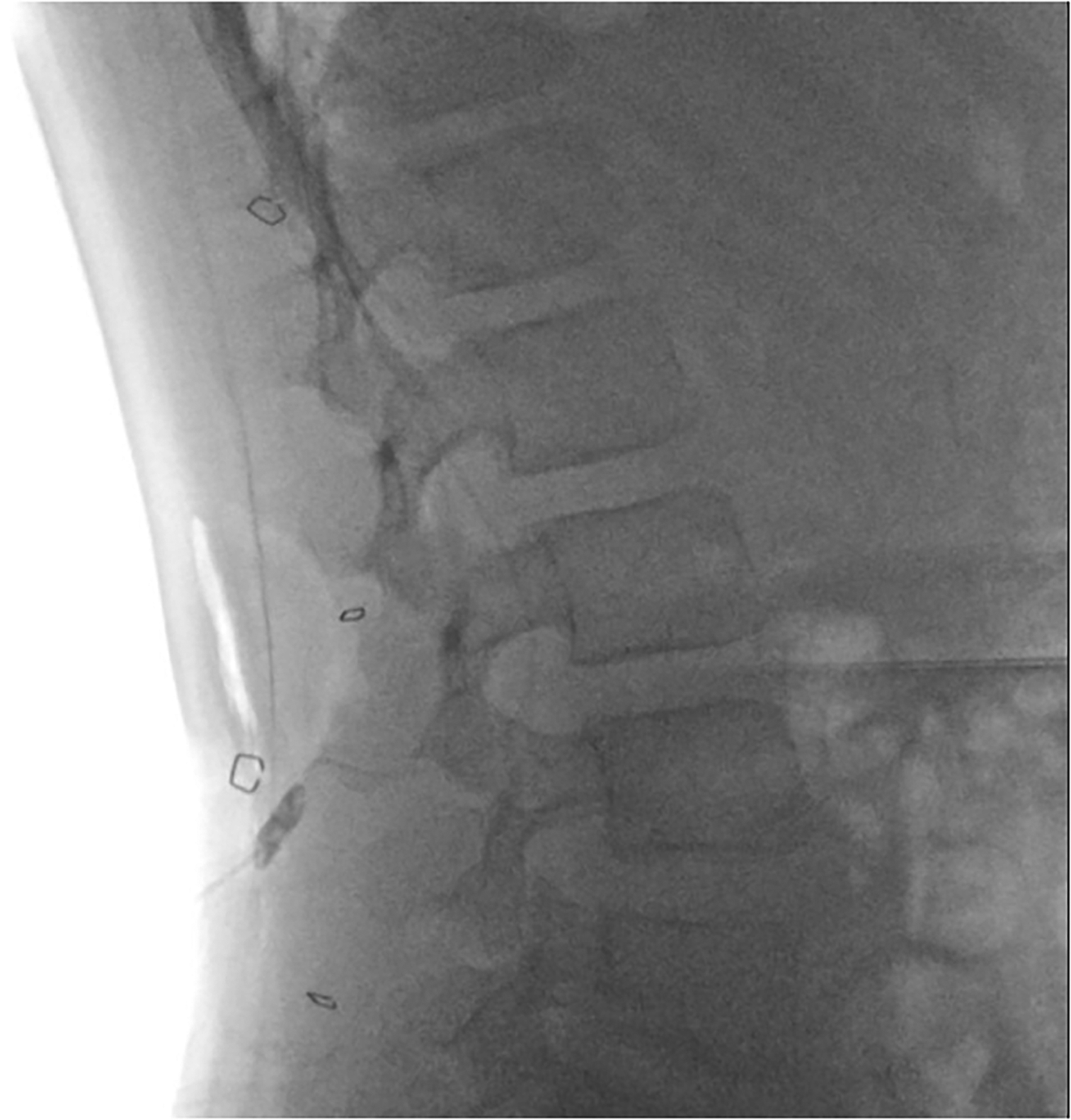
Post-operative fluoroscopy. Fluoroscopic image demonstrating a successful placement of the intrathecal catheter in the lumbar thecal sac.
Once position is confirmed, the introducer needle and stylet are withdrawn while maintaining the intrathecal position of the catheter. Patency is verified by cerebrospinal fluid (CSF) outflow and fluoroscopy verifies an unchanged catheter position. A purse-string suture at the dural entry site can be placed to reduce leaks if an open laminectomy is performed. The catheter is then anchored at the fascial or subfascial entry point with the dedicated fixation device, which is secured to fascia with interrupted non-absorbable sutures. When feasible, the fixation device is placed on the underside of the fascia with a fascial closure above it.
The catheter is subsequently tunneled subcutaneously or subfascially to a port pocket created by blunt dissection. Port site selection allows comfort and anatomical considerations, aiming to minimize pressure from the patient's bed or wheelchair. After tunneling, the proximal catheter is trimmed to allow both a 2 cm insertion segment into the port connector and 15 cm of slack for strain relief to accommodate growth and movement. The catheter-port connection is secured with the connector thumbscrew and torque wrench. Patency of the assembled system is re-confirmed by aspiration of CSF or fluoroscopic contrast injection, followed by a sterile saline flush.
The port is then anchored to underlying tissue with non-absorbable sutures at four points and the strain-relief loop is positioned underneath or remote from the port to avoid catheter migration. After ensuring hemostasis, the pocket and incision sites are irrigated and closed in anatomical sequence. Sterile dressings are applied, and an implant card documenting patient identifiers, implant details, and port location is filled.
Drug administration procedures
For this study, subjects were scheduled for their first system access within two weeks of implantation. Procedures were performed by neurosurgeons or orthopedic surgeons experienced in intrathecal access and indwelling port placement. Sedation was usually not required for port access. Ports were accessed using sterile technique with full barrier precautions. Access was obtained using a 19-to-22-gauge non-coring needle advanced until it engaged the silicone septum. After access was confirmed, the system was flushed with 0.2 mL of normal saline to establish patency, and Nusinersen was then administered over 1–3 min according to the standard dosing protocol. After nusinersen administration via the port, the system was flushed with 3 mL normal saline to clear residual drug from the port and catheter.
Outcomes and statistical analysis
The main outcomes of this interim analysis consisted of 1) the rate of successful implantation, defined as intraoperative confirmation of correct catheter and port placement and 2) postoperative proof of device patency, defined as a successful port aspiration and Nusinersen infusion. Secondary outcomes included failure-free device survival, duration of anesthesia, total procedure time, fluoroscopy time, cumulative radiation dose, need for perioperative blood transfusion, and length of hospital stay. We report the number of subjects who reached one year of follow-up and retained a functional device at that visit. In addition, time to device action (defined as revision, replacement, or explantation) was assessed in all subjects using Kaplan–Meier analysis with right censoring, presenting the estimated probability of remaining free from device action at 90, 180, and 365 days.
Adverse events were recorded by an independent clinical reviewer regardless of suspected causality and adjudicated by a multidisciplinary safety committee. Each was categorized as serious (SAEs) or non-serious (AEs) and further classified by presumed etiology as device-related, procedure-related, or unrelated. Determination of device-relatedness required supporting evidence from imaging findings, intraoperative observations, or adjudication consensus. Multiple events per subject were permitted and reported accordingly.
Finally, a time-to-event analysis was performed to estimate the probability of remaining free from any device action using the Kaplan–Meier method with right censoring. Device actions were defined as either explantation or device replacement. Estimates of freedom from device action with 95% confidence intervals were reported at 90, 180, and 365 days. The number of patients contributing follow-up at each time point was displayed below the survival curve.
All data are summarized descriptively, with no formal hypothesis testing. Continuous variables are reported as mean or median with standard deviation (SD) or interquartile range (Q1—Q3) as appropriate. Categorical variables are summarized as counts and percentages (n, %). All analyses were conducted in RStudio (Posit PBC, Boston, MA).
Results
The first 25 subjects recruited at five different sites for the PIERRE trial were included in this report. Mean age at intervention was 13.8 years (SD 5.6; range 5–22) and 13/25 (52%) were female. Body mass index ranged from 13 to 38 kilograms per square meter. Twenty-four subjects were classified as non-naïve to Nusinersen. Eleven subjects (44%) reported having respiratory comorbidities, ranging from mild respiratory disease to chronic respiratory failure; however, none were receiving mechanical ventilation at the time of implantation (Table 1). All subjects underwent an open procedure. Nevertheless, in 20 patients the lumbar spine was not exposed, as the lumbar catheter was introduced transfascially using a needle to access the thecal sac. In the remaining five subjects, the dura was surgically exposed via laminectomy.
Baseline characteristics.

Continuous variables summarized as mean with standard deviation or median with interquartile range, as appropriate. Categorical variables reported as count over total with percent.
All 25 (100%) planned and attempted procedures were successful. As of the data cutoff for this interim analysis, 57 Nusinersen infusions have been successfully delivered via the port in 23 of 25 subjects (92%). Among them, 6 had received one maintenance dose, 6 had received two maintenance doses, and 11 had received three maintenance doses. Median follow-up was 230 days [76–369] and median time from implantation to first access was 14 days [8–45]. Ten patients reached one year of follow-up, and all maintained a functional device at this visit.
All surgical implantations were performed under general anesthesia. Intraoperative imaging was routinely used to verify intrathecal position and patency prior to closure: fluoroscopy alone in 22 (88%), radiographs alone in 2 (8%) and both in 1 (4%). Median fluoroscopy time was 36 s [30–56.8] and the median intraoperative radiation dose was 6.0 mGy [2–16.1]. Perioperative transfusion occurred in a single subject (4%) during concurrent spinal deformity correction surgery. Median length of hospital stay was 1 day [0–2], and one hospitalization of 12 days occurred in one of the subjects who underwent concurrent posterior spinal fusion (Table 2).
Summary of secondary outcomes according to procedure category.

Implantation only denotes port catheter placement without other surgery. Implantation and surgical procedure denotes concurrent spine operation. All values are medians with interquartile ranges.
A total of 60 adverse events occurred in 19 subjects, with 30 events (50%) occurring within 30 days post-implant. None of the events were classified as unanticipated, and a summary of all registered events is provided in the Supplementary Content 1. Among all events, 12 events (20%) were adjudicated as serious adverse events (SAEs). The most frequently reported SAEs were wound-related complications (n = 3, 5%) and upper respiratory infections (n = 2, 3.3%). One SAE (1.6%) was adjudicated as device-related, involving excessive granulation tissue at the port site, which developed 227 days after implantation. Among the remaining SAEs, 4 (6.6%) were adjudicated as procedure-related, while 7 (11.6%) were considered unrelated. Procedure-related SAEs were limited to inability to aspirate cerebrospinal fluid, wound dehiscence, wound drainage, and implant site swelling. No SAEs were attributed to the drug itself.
The remaining events (n = 48, 80%) were classified as non-serious adverse events (AEs). Of these, 24 events (40%) were deemed procedure-related, while the remaining 24 (40%) were considered unrelated to either the procedure or the device. Among the procedure-related AEs, six (10%) were dermatologic reactions and six (10%) were related to the wound or device implantation site. The most frequently reported unrelated AE was respiratory tract infection, with nine (15%) occurrences. No non-serious events were attributed to Nusinersen administration.
Figure 3 summarizes time to device action using a Kaplan–Meier estimate, with the numbers below the curve showing participants contributing follow-up at each time point. At 90 and 180 days, the estimated probability of remaining free from device action was 91.6% (95% CI 0.81–1.00), and at 365 days it was 86.2% (0.73–1.00). One postoperative device replacement was performed due to catheter migration. Two of the devices were explanted. The first was a full system explantation prompted by wound dehiscence and skin breakdown in the setting of incomplete adherence to wound care instructions. The second was a port-only removal due to persistent access difficulties, which ultimately required revision surgery and replacement. These two events were the only AEs or SAEs that resulted in hospitalization during the study follow-up period.
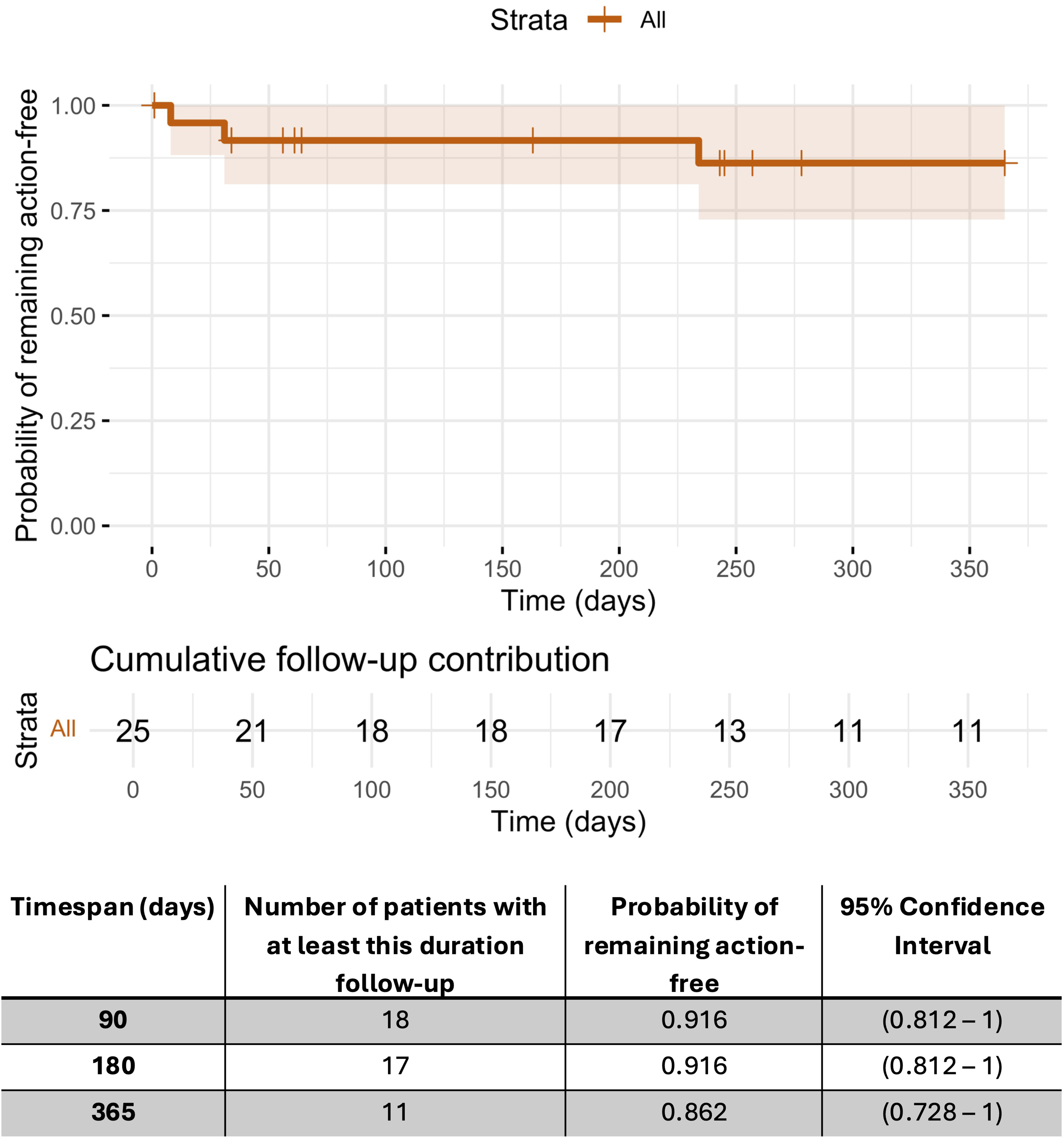
Kaplan-Meier estimate of freedom from device-action. The step curve shows the probability of remaining free from any device action, defined as any device related procedure including revision, replacement, or explantation. The shaded band represents the 95 percent confidence interval. Numbers displayed beneath the curve indicate the count of participants contributing follow-up at each time point.
Discussion
In this preliminary feasibility report, implantation of a dedicated catheter–port system for intrathecal Nusinersen was technically successful in all 25 subjects, and 23 retained the device and received at least one infusion. Overall, these results, which align with other reports describing high implantation success using custom-assembled configurations,15,16 suggest that a dedicated catheter–port system can be implemented consistently as a permanent intrathecal access option across multiple centers caring for patients who live with SMA.
Because limiting enrollment to patients with a low comorbidity burden would produce a sample unreflective of the full intended target population, the PIERRE-IDE study included individuals with prior spinal instrumentation and baseline pulmonary impairment. In this representative study population, outcomes were consistent with a straightforward intervention; median hospitalization was 1 day, and most subjects were discharged by postoperative day 2, reflecting a shorter length of stay than the average of 5 days reported for low lumbar Ommaya reservoir placement. 17 Additionally, stratified summary statistics demonstrated that standalone device implantation was characterized by a brief procedure and anesthesia, whereas longer durations occurred in concurrent spinal procedures. Prior studies assessing intrathecal device implantation have described skin-to-skin times ranging from 60 to 90 min.15,16 This modestly longer median procedure duration likely reflects additional steps for imaging confirmation and port fixation inherent to the ThecaFlex workflow, rather than increased procedural complexity.
Of note, as of the time of this report, no device has been FDA-approved for repeated intrathecal Nusinersen administration. Most prior alternatives have adapted catheters and ports intended for other indications into a composite, off-label system.9,11,15 While creative, this strategy adds interfaces and may complicate troubleshooting. To date, only one report has described a purpose-built system designed specifically for repeated intrathecal access; this device was evaluated in eight patients and suggested feasibility, although instances of device leakage were reported. 16 Additionally, suboccipital or dorsal implantation at varying levels has been explored, mainly based on theoretical considerations related to CSF flow dynamics, segmental drug exposure, or to address cases in which lumbar anatomy is altered by spinal deformity.12,15 Lumbar implantation, however, offers practical advantages, as it is a well-established approach and allows the port to be positioned in a less conspicuous location compatible with routine activities, including wheelchair use and seated transfers.
While pulmonary and anesthesia-related AEs were not prespecified, the considerable comorbidity burden in patients with SMA merits careful contextualization of these events. 18 No anesthesia-related SAEs were observed in this initial group of patients and respiratory-related AEs occurred in a subset of patients. Notably, these events were generally delayed relative to implantation and did not require hospitalization, suggesting a limited temporal association with the perioperative period. These preliminary data are consistent with prior reports describing a low incidence of anesthesia-related complications in SMA (<5% in pre–disease-modifying therapy cohorts)19,20 and supports the notion that, in patients living with SMA who do not have clear contraindications to surgery, device implantation requiring general anesthesia is usually safe.
Finally, results appear consistent with expectations for a spinal access procedure in this population, with no reported cases of bacterial meningitis or CSF leak—complications that warrant proactive monitoring from healthcare providers and investigators. The three serious wound-related AEs are more plausibly technique- and wound-care related than intrinsic device failure; given the generally low BMI in this cohort, careful soft-tissue handling, and pressure-minimizing precautions are particularly important. Accordingly, optimization should target both intraoperative technique and postoperative management, including standardized port fixation workflows, appropriate dressing selection, routine port-site surveillance, and structured caregiver education in wound care. With recruitment expanded to 20 active sites and a target enrollment of 90 participants, the definitive report of the PIERRE-IDE study will provide a more robust assessment of device performance, clarifying the distinction between technique-sensitive wound events and later outcomes informative of device durability and long-term functionality.
Conclusions
The ThecaFlex DRx system is a dedicated catheter–port designed to address previously reported limitations of intrathecal drug delivery in patients with SMA. Across participating centers, implantation was technically successful and generally well tolerated. Hospital stays were relatively short, and AEs were predominantly procedure related. As the PIERRE-IDE study continues, these preliminary findings support the feasibility and an acceptable early safety profile of ThecaFlex DRx as a potential delivery option for nusinersen.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602261432303 - Supplemental material for Clinical evaluation of ThecaFlex DRx, a novel implantable catheter-port for intrathecal nusinersen delivery in spinal muscular atrophy: Initial results from the PIERRE–IDE study
Supplemental material, sj-docx-1-jnd-10.1177_22143602261432303 for Clinical evaluation of ThecaFlex DRx, a novel implantable catheter-port for intrathecal nusinersen delivery in spinal muscular atrophy: Initial results from the PIERRE–IDE study by Ignacio Mesina-Estarrón, Michael Abruzzo, Fernando Cotrim Gomes, Melina Thaxton, Aaron Yengo-Kahn, Scellig Stone, Weston Northam, Brian Snyder, Patrick Cahill, David Bauer and Michael Muhonen in Journal of Neuromuscular Diseases
Supplemental Material
sj-doc-2-jnd-10.1177_22143602261432303 - Supplemental material for Clinical evaluation of ThecaFlex DRx, a novel implantable catheter-port for intrathecal nusinersen delivery in spinal muscular atrophy: Initial results from the PIERRE–IDE study
Supplemental material, sj-doc-2-jnd-10.1177_22143602261432303 for Clinical evaluation of ThecaFlex DRx, a novel implantable catheter-port for intrathecal nusinersen delivery in spinal muscular atrophy: Initial results from the PIERRE–IDE study by Ignacio Mesina-Estarrón, Michael Abruzzo, Fernando Cotrim Gomes, Melina Thaxton, Aaron Yengo-Kahn, Scellig Stone, Weston Northam, Brian Snyder, Patrick Cahill, David Bauer and Michael Muhonen in Journal of Neuromuscular Diseases
Footnotes
Acknowledgements
We thank the patients and their families for their participation in this study, as well as the clinical and research staff for their support during the execution of this study.
Ethical considerations
Institutional review board approval was obtained at each participating site prior to enrollment. IRB committees included those at CHOC, Boston Children's Hospital, Texas Children's Hospital, and the Children's Hospital of Philadelphia. Written informed consent was obtained from a parent or legal guardian for each minor participant, with assent obtained when appropriate.
Author contributions
All authors contributed to critical revision of the manuscript, approved the final version, and agreed to be accountable for all aspects of the work.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Alcyone, Inc,
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data generated and analyzed in this study are proprietary. Inquiries regarding data access or transparency should be directed to the corresponding author.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.