
Abstract
Haploinsufficiency of SOX5 causes Lamb–Shaffer syndrome, a rare condition with developmental delay, impaired language and intellectual disability and optic nerve abnormalities. Muscle involvement is poorly characterized. We report a 20-year-old woman with neonatal hypotonia, delayed motor milestones, proximal weakness and learning difficulties. Serum creatine kinase was normal. Electroneuromyography revealed a mild myogenic pattern. Muscle biopsy revealed myopathic changes, with cores and protein aggregates. Whole genome sequencing disclosed a heterozygous pathogenic variant in the SOX5 gene. This case expands the phenotypic spectrum of Lamb-Shaffer syndrome, confirming primary muscle involvement with core myopathy.
Introduction
The SOX gene family includes over 20 genes involved in development, cell fate, and tissue differentiation.1–3 Pathogenic variants in SOX genes have been linked to a variety of congenital diseases. 3 Among them, SOX5, on chromosome 12p12.1, encodes a transcription factor critical for neural development, chondrogenesis, spermatogenesis and tumorigenesis.1–4 Heterozygous pathogenic variants in SOX5 cause Lamb–Shaffer syndrome (LAMSHF, MIM 616803), a neurodevelopmental disorder with developmental delay, language and intellectual impairment, behavioral disorders and distinct facial features.3,5–14 Although hypotonia, muscle weakness, and decreased muscle bulk have been reported, muscle involvement remains poorly characterized.5–14 Here, we describe for the first time the detailed clinical and myopathological features of a 20-year-old woman with early-onset muscle weakness, exercise intolerance and a heterozygous pathogenic variant in SOX5.
Methodology
The patient was examined by one of the authors at the Neuromuscular Reference Center and Genetics Department at Pitié Salpêtrière and Trousseau Hospital in Paris, France. The patient provided written informed consent to participate in the study and for publication. The study was conducted in compliance with European Union and French bioethics laws, as well as the Declaration of Helsinki.
Electroneuromyography (ENMG), blood tests, brain and muscle MRI results were performed. Muscle biopsies were processed for standard histological and immunohistochemical studies, and glutaraldehyde-fixed for electron microscopy. Trio-based whole-genome sequencing (WGS) was performed at the “Laboratoire de Biologie Médicale Multi Sites SeqOIA” (https://laboratoire-seqoia.fr/). DNA was extracted from blood cells. Libraries were prepared using the Illumina DNA PCR-free Prep, Tagmentation protocol. Paired-end (2 × 150) sequencing was performed on Flow Cell S4, NovaSeq 6000®, Illumina®. Raw output was demultiplexed (bcl2fastq®, v2.20.0.422, Illumina®) and aligned on the GRCh38 reference genome using BWA-MEM2 (v2.2.1). Duplicates were marked using Picard MarkDuplicates (2.8.1), base quality was recalibrated with GATK4 (v4.1.9.0, Broad Institute). Small variants were called using GATK4 (v4.1.9.0, Broad Institute) and annotated by SNPeff (v4.3t). Structural variants were called with ClinSV (1.0.1) and WiseCandorX (v1.2.4) and annotated by AnnotSV (v3.0.7). An average depth-of-coverage of 37x was obtained for the proband and the parents. Variants were prioritized according to ACMG guidelines. 15
Case presentation
A 20-year-old Caucasian woman was referred for suspected congenital myopathy due to delayed motor milestones, exercise intolerance, and myalgia.
She was born at 36 weeks from a bi-chorionic, bi-amniotic pregnancy complicated by preeclampsia. Her twin sister was asymptomatic, and the parents are healthy, non-consanguineous, with no neuromuscular family history.
Neonatal hypotonia and difficulties with feeding and sucking were noted, with motor delay (head control acquired at 6 months, sitting at 9, walking at 23). She also had a language delay, trouble in word articulation and learning difficulties with fine motor skill disorder. No developmental regression was noted. She attended adapted schooling from age 8. Since childhood, the patient exhibited an introverted personality with a certain degree of social anxiety. She started a vocational training program but did not complete it successfully and did not obtain a diploma. Since then, she has been working in a sheltered employment center, adapted for individuals with disabilities. At age 24, neuropsychological assessment (WAIS-IV) revealed a moderate intellectual disability, with a full-scale IQ of 46 (VCI 59, PRI 54, WMI 58, PSI 55).
Non-progressive mild proximal weakness with bilateral scapular winging was observed since infancy. Dorsal scoliosis was diagnosed at the end of the first decade of life, initially managed with a corrective brace and requiring posterior vertebral arthrodesis at age 15 (Figure 1). A flattened thorax was also present. Ophtalmological examination, including optical coherence tomography (OCT) and retinal nerve fiber layer (RNFL) analysis, revealed bilateral optic atrophy with papillary excavation, without any clinical or functional impact on the patient, who maintains normal visual acuity. There was no history of epilepsy.
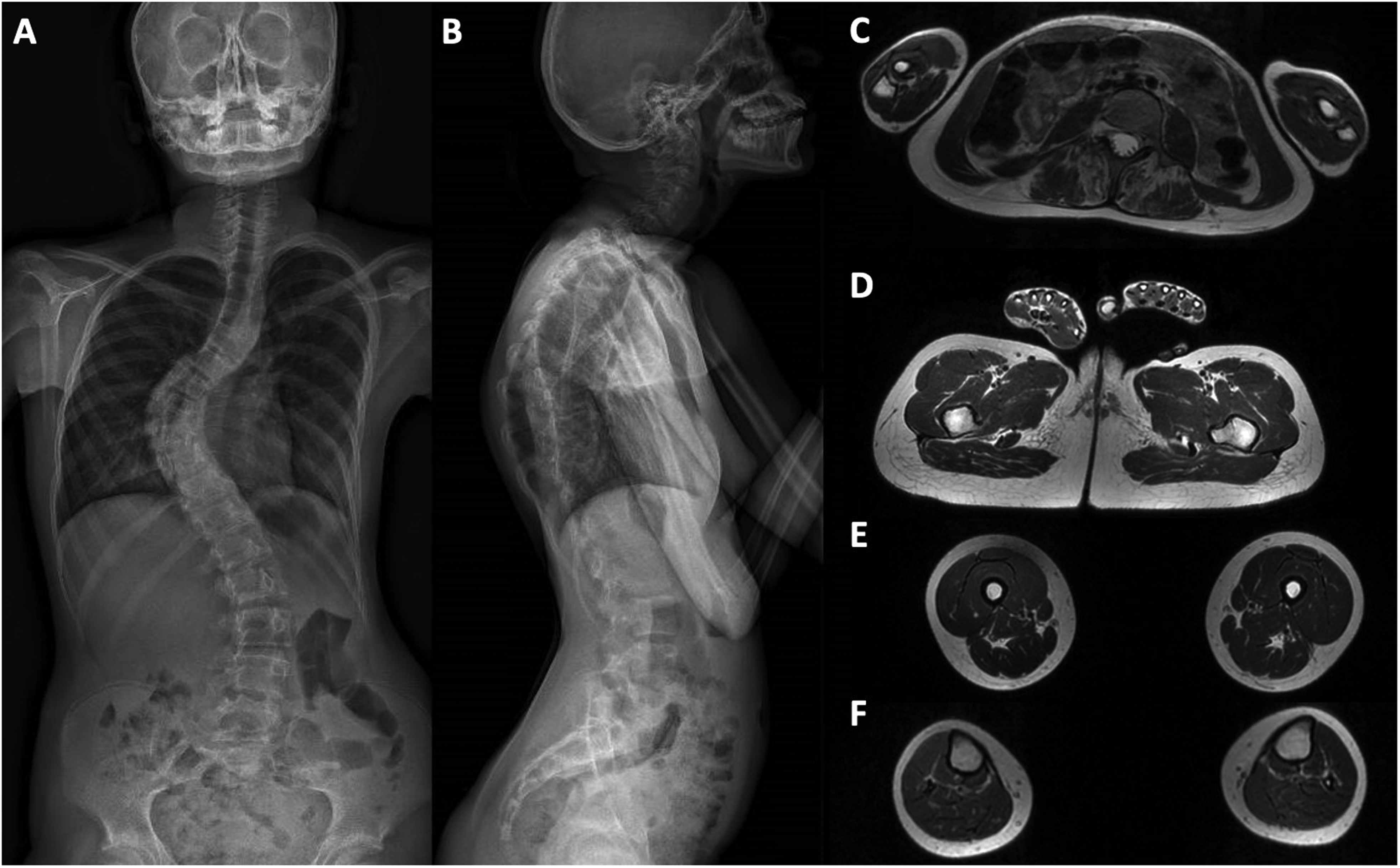
Electronic Optical Scan (EOS) X-ray imaging of the full-spine and muscle MRI. A-B: EOS X-ray imaging of the full-spine imaging shows severe scoliosis and prominent dorsal kyphosis. C-F: muscle MRI (axial, T1-weighted images) reveals severe atrophy and fatty infiltration of paraspinal muscles (C), and normal muscle signal at pelvic (D), thigh (E) and leg (F) levels.
At 20 years when we first assessed the patient, she had normal but low-range anthropometric measurements, with a height of 156 cm (−1.3 SD, target height 177 cm, +2.5 SD), a weight of 47 kg (BMI 19 kg/m2), and a head circumference of 53.5 cm (−1.3 SD). Neurological examination revealed global amyotrophy with axial and symmetrical proximal muscle weakness, with the following Medical Research Council scores: deltoid 4+/5, pectoralis major 4+/5, triceps brachialis 5-/5, psoas 4+/5, and gluteus maximus 4+/5. There was no facial weakness, ptosis, or ocular palsy. Mild finger hyperlaxity was also observed. She displayed distinct facial features, including a broad and full nasal tip, prominent upper incisors, and a small chin. Anxious behavior noted during the history taking and physical examination was confirmed by the patient's mother, who stated that this has been a longstanding personality trait since early childhood.
Electroneuromyography (ENMG) revealed normal nerve conduction studies, while electromyography (EMG) showed a diffuse mild myogenic pattern with brief and small motor unit potentials without spontaneous activity. Whole-body muscle MRI performed at age 15 revealed atrophy and fatty infiltration affecting paravertebral muscles (Figure 1), in the context of severe kyphoscoliosis, without any signal abnormalities nor muscle atrophy affecting upper or lower limbs.
The patient underwent two muscle biopsies. A first deltoid muscle biopsy performed at 7 years showed the presence of a globally preserved structure (Figure 2A), confirmed with ultrastructural electron microscopy studies (Figure 2C). A second muscle biopsy performed at 14 years showed abnormalities consistent with a congenital core myopathy. The hematoxylin and eosin (H&E) revealed the presence of central areas of the fibers with accumulation of eosinophilic material (Figure 2D); the latter corresponded to well-delimited centrally located areas of uneven oxidative staining (Figure 2E). No fatty infiltration was detected, as well as congenital type 1 fiber disproportion. In contrast there was fiber size variation and type II fiber predominance. Electron microscopy showed the presence of accumulation of Z-line material, loss of the sarcomeric structure and absence of mitochondria corresponding to core lesions (Figure 2F). Strikingly, inside the cores lesions there was the presence of dense granulofilamentous protein material (Figure 2G). Immunohistochemical studies using desmin and αB-crystallin antibodies confirmed that granulofilmaentous material was composed of desmin (Supplementary Figure 1), as has been previously described in core-myopathies. 16 The menadione alpha-glycerophosphate reaction did not reveal the presence of reducing bodies.
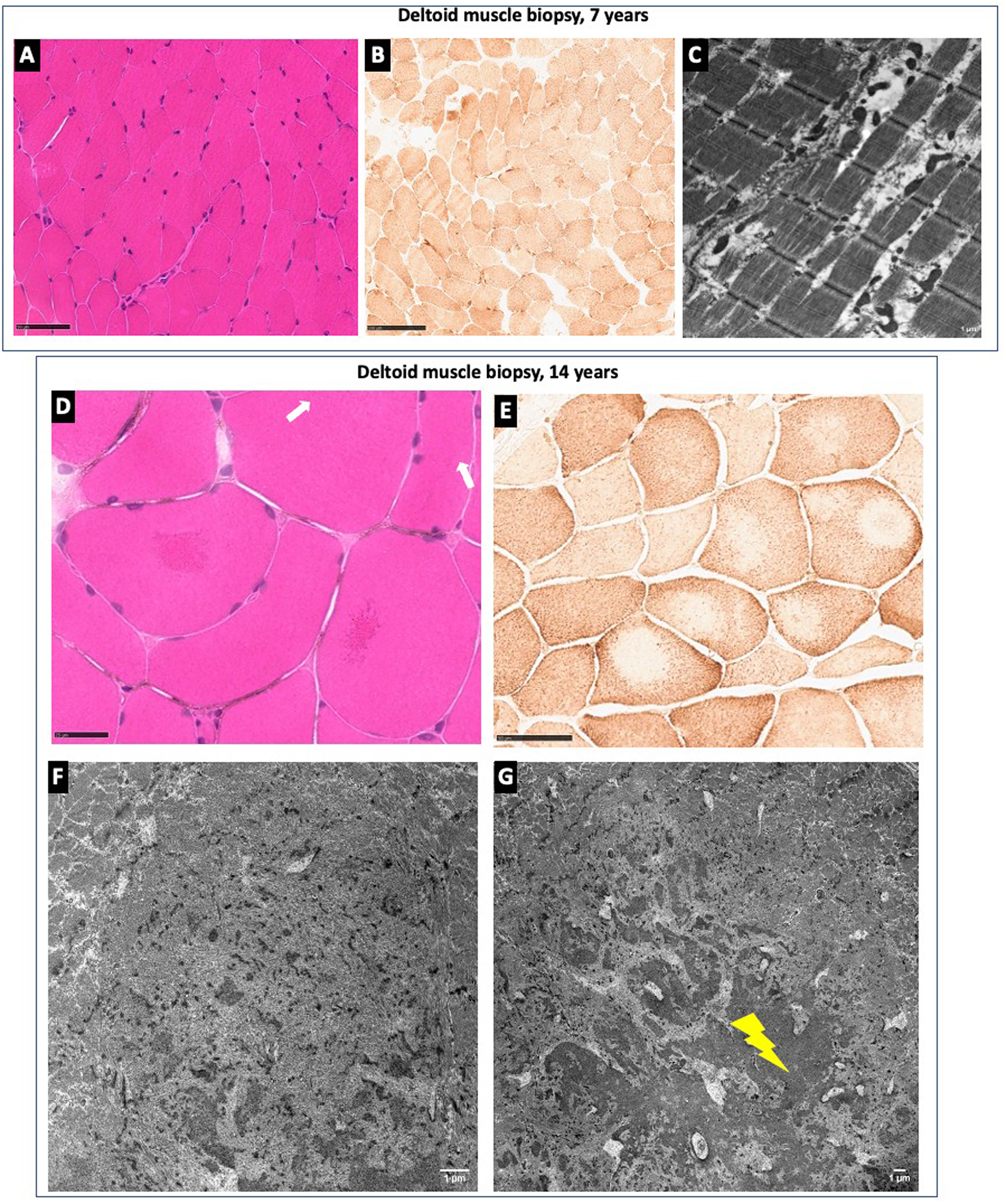
Muscle biopsy findings from deltoid muscle biopsies performed at 7 years in A, B, and C and at 14 years in D, E, F and G. A: H&E reveals mild fiber size variation. B: COX histoenzymatic reaction demonstrates increased subsarcolemmal reaction in some fibers. C: Electron micrograph illustrates mild proliferation of morphologically normal mitochondria. D: H&E highlights two fibers harboring centrally located eosinophilic material (white arrows). E: COX histoenzymatic reaction displays centrally located areas of the fibers with uneven oxidative staining. F: Electron micrograph shows a well delimited area of progressive Z-line fragmentation, accumulation of Z-line material, loss of the sarcomeric structure and absence of mitochondria corresponding to a core lesions. G: Dense granulofilamentous protein material (indicated by the yellow flash) is found associated with the core lesion.
Cardiological investigations were normal. Spirometry showed a restrictive pattern with a vital capacity at 60% of the predicted value, without any abnormalities in the arterial blood gas test. CK levels were normal. Brain MRI showed no abnormalities.
Genetic investigations, including Array Comparative Genomic Hybridization (CGH), karyotype analysis and fragile X testing were normal. Given the clinical history with delayed motor milestones and slowly progressive weakness, along with the muscle biopsy findings, a next-generation sequencing (NGS) panel analysis for congenital myopathies was performed, revealing a heterozygous variant of uncertain significance, c.13509A > C (p.K4503N) in the RYR1 gene. Segregation analysis showed that the variant was inherited from her asymptomatic mother. There was no personal or family history of malignant hyperthermia. Finally, trio WGS analysis revealed a de novo heterozygous pathogenic variant in the SOX5 gene (NM_006940.6:c.1342 + 1_1342 + 4delinsATAT), located at a canonic splicing site and predicted to abolish a donor splicing site. The variant was absent from population databases (GnomAD v4), unreported in Clinvar and in the literature, and was classified as pathogenic according to ACMG guidelines (PV1, PS2, PM2 Supp).
Discussion
Here, we describe the first detailed clinico-histological characterization of muscular involvement associated with a de novo heterozygous SOX5 pathogenic variant. The patient presented the classical phenotype associated with LAMSHF, including global developmental delay, intellectual disability, papillary excavation and distinct facial features.5–14,17 She also exhibited symptoms and signs overlapping those of congenital myopathies, such as delayed motor milestones, slowly- or non-progressive proximal muscle weakness and scoliosis. Needle EMG confirmed muscle involvement and the second muscle biopsy revealed morphological abnormalities consistent with a congenital core myopathy, namely fiber size variation along with sarcomeric disorganization and structural alterations (i.e., Z-line fragmentation, accumulation of Z-line material, loss of sarcomeric structures and areas devoid of mitochondria and oxidative activity corresponding with core lesions). Notably, ultrastructural studies revealed the presence of granulofilamentous material, consistent with desmin on immunohistochemistry. The aggregation of dense material has been previously described in another congenital myopathy, the KBTBD13-Related Congenital Myopathy. 18 Further studies are warranted to disclose the link between SOX5, myofibrillar disorganization with core areas formation and protein aggregation. It is important to note that both muscle biopsies were taken from the same muscle seven years apart. Indeed, we might speculate that the progression of the disease led to the appearance of more striking structural abnormalities in the second biopsy, particularly regarding sarcomeric disorganization, whereas the first biopsy showed only mild, nonspecific changes.
Muscle involvement in LAMSHF is commonly reported as nonspecific hypotonia, muscle weakness or decreased muscle bulk.5–14 Along these lines, a long isoform of SOX5 is expressed in skeletal muscles, explaining the potential muscle involvement.5,19 Although more than 40 patients with possible muscle involvement have been reported (Supplementary Table 1), a primary myopathic process has not been confirmed beyond the clinical impression based on symptoms and examination, and its phenotypic and histological characterization remains unexplored. Regarding orthopedic abnormalities, neuromuscular involvement may potentially contribute to the development of scoliosis, which is a common feature in congenital myopathies. 20
It should also be noted that our patient carried a heterozygous RYR1 variant of unknown significance, likely unrelated to her condition, as her unaffected mother shares the same variant, and there is no history of malignant hyperthermia in either the proband or her family.
Interestingly, mutations in another SOX gene, SOX8, have been linked to a congenital nonprogressive myopathy. Warman et al., reported a patient with short stature, skeletal dysplasia, respiratory involvement, intellectual delay, contractures, and amenorrhea, due to compound heterozygous SOX8 variants. In this case, muscle biopsy showed fiber size variation and type II fiber predominance, an atypical finding for congenital myopathies. 21 Also in patients with NEB-related nemaline myopathy, a subgroup of patients do not reveal type 1 fiber predominance, probably associated to different myosin overexpression in congenital muscle disorders. 22
Our results provide new insight into the muscular manifestations of SOX5 variants, presenting here as a congenital myopathy in association with LAMSHF's typical neurodevelopmental and ophthalmologic involvement and distinct facial features. This observation also broadens the genetic spectrum of congenital myopathies, suggests that SOX genes should be considered in congenital muscle diseases, in particular when there are associated systemic and developmental features. Moreover, it highlights the utility of WGS in complex phenotypes where targeted genetic panels for congenital myopathies are inconclusive.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602261428296 - Supplemental material for Congenital core myopathy linked to SOX5: Expanding the phenotypical spectrum of Lamb-Shaffer syndrome
Supplemental material, sj-docx-1-jnd-10.1177_22143602261428296 for Congenital core myopathy linked to SOX5: Expanding the phenotypical spectrum of Lamb-Shaffer syndrome by Katia Staedler, Anna Gerasimenko, Caroline Nava, Delphine Heron, Elodie Schaerer, Cyril Gitiaux, François-Jerôme Authier, Tanya Stojkovic, Edoardo Malfatti and Rocio-Nur Villar-Quiles in Journal of Neuromuscular Diseases
Footnotes
Acknowledgments
The authors have no acknowledgments to report.
Ethical considerations
The study was conducted in accordance with European Union and French bioethics laws and with the Convention of Helsinki.
Consent to participate
The patient gave his informed consent to participate.
Consent for publication
The patient gave his consent for publication.
Author contributions
Katia Staedler (Conceptualization; Methodology; Investigation; Writing - Original Draft); Anna Gerasimenko (Investigation; Writing - Review & Editing); Caroline Nava (Writing - Review & Editing); Delphine Heron (Writing - Review & Editing); Elodie Schaerer (Writing - Review & Editing); Cyril Gitiaux (Writing - Review & Editing); François-Jerôme Authier (Writing - Review & Editing); Tanya Stojkovic (Writing - Review & Editing); Edoardo Malfatti (Investigation; Writing - Review & Editing); Rocio-Nur Villar-Quiles (Conceptualization; Methodology; Investigation; Writing - Review & Editing).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.