
Abstract
Background
Hereditary spastic paraplegia (HSP) is a heterogenous group of rare genetic disorders characterized by progressive corticospinal and dorsal spinal cord axonal degeneration manifesting as muscle weakness and spasticity of the lower extremities. Over 98% of solved HSP cases are caused by pathogenic variants in the nuclear DNA.
Case
We report a family carrying the m.9035T > C [p.(Leu170Pro)] pathogenic variant in the mitochondrial MT-ATP6 gene in the setting of maternally inherited, late-onset HSP. The proband (age 67 years) presented with classical, late-onset, pure HSP. Her affected daughter (age 39 years) developed late-onset, complex HSP, with asymmetrical axonal sensorimotor polyneuropathy. Her second daughter (age 46 years) carried the same pathogenic variant with high heteroplasmy but was clinically unaffected at last assessment, suggesting age-dependent or incomplete penetrance.
Summary of literature
The substitution of a leucine for a proline affects a highly conserved transmembrane helix of the subunit “a” at a key functional domain in the mitochondrial ATP synthase complex. The m.9035T > C variant has been reported in several families presenting with common phenotypic presentations of ATP6-related disorders such as maternally inherited Leigh syndrome (MILS) and the syndrome of neuropathy, ataxia, and retinitis pigmentosa (NARP). HSP is a rare presentation in ATP6-related disorders; mitochondrial ATP6-induced HSP has previously been published in only one family carrying a homoplasmic m.9176T > C [p.(Leu217Pro)] variant.
Conclusion
This report highlights the role of MT-ATP6 pathogenic variants in complex and pure HSP and raises the relevance of genetic testing of MT-ATP6 in undiagnosed cases of sporadic or maternally inherited HSP.
Keywords
Introduction
Hereditary spastic paraplegia (HSP) is a heterogenous group of rare genetic disorders characterized by progressive corticospinal and dorsal spinal cord axonal degeneration manifesting as muscle weakness and spasticity of the lower extremities. Prevalence of HSP is estimated ∼1 to 5 cases in 100,000 individuals. 1 HSP typically presents after age 35, but early onset forms (infancy, toddler, or adolescence) have been reported. HSP can be clinically categorized as “pure” or “complex.” In “pure” forms, neurologic impairment is limited to progressive lower-extremity spastic weakness, hypertonic urinary bladder disturbance and mild diminution in vibration sensation. Conversely, “complex” forms are defined by the presence of various additional manifestations such as cognitive involvement, seizures, ataxia, peripheral neuropathy, and extrapyramidal features. 2
More than 88 genes have been associated with HSP, 3 solving only half of HSP cases. 4 Autosomal dominant forms are predominant, autosomal recessive represent less than 30% of HSP cases, and X-linked recessive as well as mitochondrial inheritance represent the rarest forms, each estimated at 1–2% of all HSP cases.1,2 To date, four mitochondrial genes have been associated with HSP: MT-TI (1 family) 5 ; MT-CO3 (1 family) 6 ; MT-ND4; and MT-ATP6 (very few families).7–10 In addition, variants in at least 10 nuclear genes have been associated with mitochondrial dysfunction in HSP.2,11–13 Moreover, polymorphisms in the mitochondrial DNA may also contribute to the development of HSP or act as modifiers of the phenotype. 14 Overall, these findings highlight the key roles of mitochondrial gene variants contributing to HSP pathomechanisms.
MT-ATP6 (MIM:516060) is a 681 bp mitochondrial gene encoding for subunit “a” of the mitochondrial ATP synthase, also known complex V of the mitochondrial respiratory chain. 15 The ATP synthase is located at the inner mitochondrial membrane (IMM) and plays a key role in coupling the electron transport chain to the proton gradient, for production of ATP. The phenotypic spectrum of ATP6-related disorders is extremely broad in terms of age-of-onset, disease severity and tissue/organ involvement. The most common phenotypic presentations are maternally inherited Leigh syndrome (MILS) and the syndrome of neuropathy, ataxia, and retinitis pigmentosa (NARP), but overlaps with Charcot-Marie-Tooth (CMT) and spinocerebellar ataxia (SCA) are not uncommon.16–19 We report a family of three individuals carrying the pathogenic variant MT-ATP6:m.9035T > C, with two individuals presenting with HSP with variable expressivity. In the present work, we discuss the place of this mitochondrial gene in the diagnostic strategy for HSP.
Cases
For all three cases, investigations showed white blood cell count, hemoglobin, hematocrit, mean corpuscular volume, and vitamin B12 in the normal range. Results of additional investigations are listed within each case description.
Case 1
The proband is a 67-year-old woman who presented to the Neuromuscular clinic with complaints of progressive gait ataxia. She was born in Chile of non-consanguineous parents. She first noted lower extremity stiffness at age 64, without focal muscle weakness, and began using a walker at age 66. However, her family reports very subtle gait difficulties as far back as her late 30's. She denied any cognitive changes, dysphagia, and bladder or bowel control issues. The pedigree of this patient is shown in Figure 1(a). The patient's parents were unaffected and passed in their 80 s. She has had two daughters, one clinically unaffected (case 2) and one affected (case 3), and the major features of these three individuals are summarized in Table 1.

The m.9035T > C (Leu170Pro) variant in relation to other pathogenic and likely pathogenic variants reported in the MT-ATP6 gene. (a) The pedigree of the present proband (arrow, case 1) and affected relatives (cases 2 and 3) with the MT-ATP6 gene mutation is illustrated. (b) Alignments of partial sequence of MT-ATP6 from multiple species, performed with www.mitomap.org. The arrow indicates the leucine residue (L) at position 9035. Human (Homo sapiens): Q13148, chimpanzee (Pan troglodytes): H2PY00, mouse (Mus musculus): Q921F2, cow (Bos taurus): G3MX91, horse (Equus caballus): F6WAU6, platypus (Ornithorhynchus anatinus): F7EDX1, chicken (Gallus gallus): Q5ZLN5, frog (Xenopus laevis): A0A8J0TE74. (c) Predictive tools of variant pathogenicity, from www.mitomap.org. (d) Pathogenic and likely pathogenic point mutations in the MT-ATP6 gene reported in ClinVar, and their localization within the protein domains (upper) and gene (lower). Pathogenic variants are represented by square boxes, and likely pathogenic variants by round edges boxes. Phenotypes involving mainly cardiac manifestations are represented by boxes with dashed lines. Variants associated with hereditary spastic paresis (HSP) presentations are highlighted in bold. Created with BioRender.com.
Main clinical and genetic characteristics in cases 1 to 3.

NR: not reported.
Clinical examination at 67 years of age revealed a short-statured woman (height 150 cm; <3rd percentile, and weight 58.6 kg) with asymmetric bilateral spastic gait. On motor testing, tone was normal in the upper limbs and increased in the lower extremities, without muscle wasting or fasciculations. Power was MRC (Medical Research Council) scale 5 out of 5 in proximal and distal muscles of the upper and lower extremities. Reflexes were 2+ in the upper extremities, 3+ in the lower extremities; Hoffman sign was negative bilaterally. There was no clonus and plantar responses were upgoing bilaterally. Sensory testing to light touch, pinprick and vibration were normal in the upper extremities. Mild loss of vibration was noted in the great toes bilaterally. Funduscopic exam and extraocular movements were normal. There was no dysmetria or ataxia, no dysarthria, or cognitive involvement, no dysmorphia or skeletal deformities, and no cardiac involvement. One year later, the patient developed moderate bilateral lower limb weakness, especially in hip flexors (4/5) and hamstrings (4+/5) and was unable to stand on her heels. At the last follow-up, (71 years old, almost 4 years after the initial presentation), her symptoms had progressed considerably with loss of autonomy and significant muscle fatigue. She was observed to have spastic speech and mild bilateral ptosis, without restriction of extraocular movements. Spasticity and motor weakness had progressed to the upper limbs (4/5 weakness of the deltoid muscles). Sensory examination remained unchanged. She was mildly dysmetric with finger–nose maneuver and had considerable gait ataxia. She required assistance to walk.
Investigations showed normal creatine kinase (CK) (49 U/L, reference 39–192 U/L), elevated blood lactate (5.0 mmol/L, reference 0.5–2.5 mmol/L), elevated blood alanine (731 umol/L, reference 200–483 umol/L) with elevated blood alanine:lysine ratio (4.1), and elevated Growth Differentiation Factor 15 (GDF15; 2960 pg/ml, reference <750 pg/ml). Vitamin E, copper, and red blood cell count were all in normal limits. Apart from a remote infarct in the right anterior basal ganglia, brain and spine MRI were unremarkable. Nerve conduction studies showed no convincing evidence of polyneuropathy (normal motor and sensory amplitudes and velocities in the upper and lower limbs).
Genetic testing included an autosomal dominant HSP gene panel (The Hospital for Sick Children, 2018) which identified a heterozygous variant of unknown significance (VUS) in the TECPR2 gene [c.1643A > G, p.(Asn648Ser)] that was absent from the patient's affected daughter (case 3). There was no deletion or duplication of TECPR2 and no second variant identified. This VUS had been previously associated with recessive forms of HSP and thought not likely contributing to the phenotype. There was no expansion in the C9orf72 gene. Following non-diagnostic clinical testing, this individual and her clinically affected daughter were subsequently enrolled in the Care4Rare Canada research program; study design approval was obtained from the institutional research ethics board (Children's Hospital of Eastern Ontario; CTO-1577). Research exome analysis performed in 2021 was negative. Research genome sequencing identified a missense m.9035T > C, [p.(Leu170Pro)] variant in the MT-ATP6 gene at 83.6% heteroplasmy in blood cells. RNA sequencing from the blood performed in parallel did not show outlying expression of this gene nor did it identify any other candidates. These findings were confirmed by an independent clinical laboratory. This variant was absent in the homoplasmic state in gnomAD v4.1.0 and reported twice in the heteroplasmic state (one individual listed with 0.7–0.8, and one individual with 0.92). The affected residue is highly conserved between species (Figure 1(b)). This variant was previously identified in less than 20 unrelated individuals (11 submissions to ClinVar),8,17,20–24 presenting with cerebellar ataxia, progressive gait disturbance, sensory neuropathy, and fatigue, and is classified as likely pathogenic (NICHD/NINDS U24 Mitochondrial Disease Variant Curation Expert Panel, 2022, Figure 1(c)). 25 The patient was started on a mitochondrial cocktail (Creatine 5 g daily, Ubiquinone 150 mg BID, Alpha lipoic 200 mg daily, and Vitamin E 200 IU daily) but unfortunately has continued to show clinical progression.
Case 2
The proband (case 1)'s oldest daughter is a 46-year-old female patient, born in Chile. She had a history of migraine since childhood, anxiety, depression, and post-traumatic stress disorder. She presented with symptoms of fatigue and memory difficulties. She denied dysphagia, gait disturbances, or bladder or bowel control issues. Clinical examination revealed a short-statured woman (height 147 cm; <3rd percentile, and weight 56 kg) with no gait ataxia or spasticity, no dysarthria, no dysmetria, and normal cranial nerve examination. Nerve conduction studies and needle EMG were not performed.
Given her mother's history, testing of the HSP gene panel (The Hospital for Sick Children, 2020) was performed and did not identify any variants. However, later confirmatory testing identified the same variant in the MT-ATP6 gene as the proband and case 3, present at 90% heteroplasmy in her blood cells. Red blood cell count, CK, lactate, plasma amino acid profile, urine organic acid profile, and GDF15 levels were all within the normal range. The patient was also started on a mitochondrial cocktail. Case 2's daughter has a history of asthma, anxiety, IBS, ulcers, keratoconus and astigmatism; she carries the familial variant at 25% heteroplasmy. Her son has not had genetic testing, and has a history of asthma, anxiety, and ADHD.
Case 3
The proband's clinically affected daughter is a 39-year-old female patient evaluated for spastic paraparesis, dysarthria, gaze evoked nystagmus, high arched feet, hammertoes, and distal large-fiber sensory loss. She first noticed increasing gait instability at age 35. However, her family reported issues with learning, speech, coordination, and falls since childhood. The patient denied cognitive changes, dysphagia, and bowel or bladder control issues. Clinical examination at 39 years of age revealed an average-statured woman (height 160.5 cm; 25–50th percentile, and weight 67.4 kg) with moderate dysarthria, and horizontal gaze evoked nystagmus in lateral direction. Motor examination revealed significant spasticity restricted to the lower limbs, as well as high arched feet and hammer toes. There was no motor weakness (power was MRC grade 5/5 diffusely), and no fasciculations. Reflexes were 2–3+ in the upper limbs, 3+ at the patellar tendons with positive crossed adductors, and 2+ in the ankles. The plantar responses were extensor bilaterally. She could not detect vibration at the great toes, but pinprick and light touch sensation were normal. She had mild dysmetria in the lower but not upper limbs. Her gait was spastic/scissoring with mildly impaired tandem gait. She was initially treated with low doses of baclofen.
One year later her dysarthria had worsened, and spasticity had progressed to the upper limbs. There was no motor weakness but new decreased pinprick sensation in the upper and lower limbs bilaterally were observed, as well as decreased vibration bilaterally to the left knee. Investigations showed lactate, plasma amino acid profile, urine organic acid profile, zinc, copper, vitamin E, and GDF15 levels within the normal range. CK was intermittently mildly elevated (221 U/L, reference 39–192 U/L). Nerve conduction studies showed electrophysiologic evidence of an asymmetric/multifocal sensorimotor axonal polyneuropathy (Supplemental Table). MRI of the spinal cord was normal. Genome sequencing performed alongside that of the proband identified the familial variant in the MT-ATP6 gene at near homoplasmy (99.7% heteroplasmy) in DNA extracted from white blood cells. She has two children, both minors with no genetic testing. Her daughter was reported to be clinically unaffected while her son has ADHD and is described as “clumsy” but he had not been clinically evaluated.
Discussion
Maternal inheritance is an exceedingly rare cause of HSP and one which typically results in a complex HSP phenotype. 2 We report a family carrying the m.9035T > C pathogenic variant in the MT-ATP6 gene in the setting of maternally inherited, late-onset, classic, pure HSP. This represents the first convincing description of a relatively pure late-onset HSP phenotype in medical literature with this specific variant in the MT-ATP6 gene and is associated with age-dependent or incomplete penetrance.
The m.9035T > C variant has been reported as likely pathogenic; it substitutes a highly conserved leucine (Leu) for proline (Pro) at position 170, on one of the 6 conserved transmembrane helices of the subunit “a”, at the F0 functional domain of the ATP synthase complex. This missense variant induces a change of amino acid at helix α5, a crucial spot of interaction with the c8-ring (nuclear encoded subunit “c” functional domain, which participates in proton translocation across the inner mitochondrial membrane. The flow of protons across the inner mitochondrial membrane induces a rotation of the c8-ring, thereby driving the production of ATP. 15 Altogether, the m.9035T > C variant is expected to affect the mitochondrial ATP-synthase-driven energy production. 26
There is considerable variability in the clinical phenotype associated with the m.9035T > C variant and patients mostly present with early-onset ataxia, cognitive involvement, and peripheral neuropathy, but not with late-onset pure HSP as in the present report. 8 Extensor plantar reflexes, increased muscle tone, urinary retention, and spastic gait are rarely reported. 8 A submission to ClinVar in October 2018 without referenced publication reported spasticity, spastic paraplegia, gait disturbance, difficulty walking, and fatigue in one individual.
Three other likely pathogenic and pathogenic variants of MT-ATP6 (at different sites) include features of complex HSP in their phenotypic spectrum, and are presented in Table 2. For example, late-onset HSP has been reported in five individuals carrying a homoplasmic m.9176T > C [p.(Leu217Pro)] variant, 7 affecting the distal part of the MT-ATP6 protein (Figure 1(d), Table 2). These individuals presented with spasticity and reduced vibration sense in the lower extremities. Three of them also had axonal neuropathy. Three female relatives presented with late-onset pain and spasticity of the lower limbs and one of them developed dementia, suggesting gender-related modifiers in this family. 7 In another study, an adult patient with a m.8618dup [p.(Thr33Hisfs*32)] variant was reported with developmental delay, learning difficulties, kidney failure, hearing loss, diabetes, cataract, ataxia and spastic paraparesis. 9 Two unrelated individuals carrying the m.8777T > C [p.(Leu84Pro)] variant and complex HSP have recently been published. 10 Additional variants to MT-ATP6 associated with HSP-like features have been submitted to ClinVar without accompanying publications. These variants include m.9176T > G [p.(Leu217Arg)] on June 17, 2021, m.9176T > C [p.(Leu217Pro)] on June 1, 2022 and Nov 23, 2023, m.8993T > C [p.(Leu156Pro)] on Oct 23, 2020, and m.9049G > A [p.(Gly175*)] on Dec.9, 2019. Three out of the six MT-ATP6 variants reported in ClinVar as associated with HSP clustered in the gene region coding for the helix α5 of the protein. In addition, several single-nucleotide polymorphisms associated with HSP-like phenotypes cause substitution of leucine by proline, which would destabilize the helical secondary structure 27 (Figure 1(d), Table 2). It would therefore be interesting to confirm such genotype-phenotype potential correlation in further studies.
Review of genetically confirmed cases of MT-ATP6-related disorders reported as hereditary spastic paraplegia (HSP).
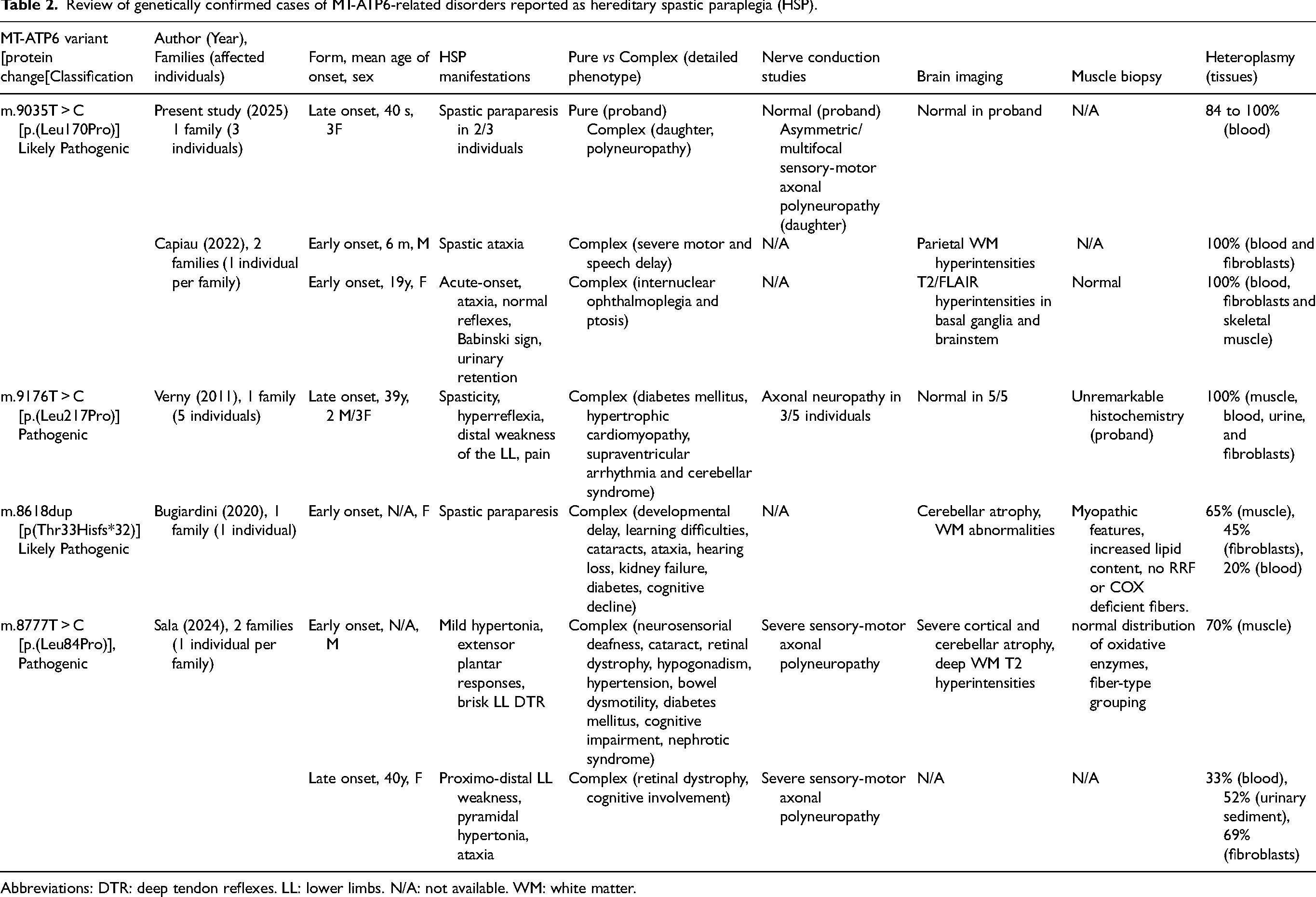
Abbreviations: DTR: deep tendon reflexes. LL: lower limbs. N/A: not available. WM: white matter.
In the present study, the proband presented with classical, late-onset, pure HSP, one daughter was clinically unaffected at the time of assessment (age 46 years), and one daughter had developed classical, late-onset, complex HSP with additional asymmetrical axonal sensorimotor polyneuropathy. Penetrance is incomplete and not explained by the mutant load/degree of heteroplasmy measured in the blood (which is not indicative of heteroplasmy level in other tissues such as neurons), as previously reported. 7 Asymptomatic patients with pathogenic MT-ATP6 variants are described in ATP6-related disorders. 17 While the proband displayed normal nerve conduction studies, the clinically affected daughter presented with asymmetric/multifocal sensorimotor axonal polyneuropathy. This asymmetric presentation is quite unusual. In the literature, MT-ATP6 variants have been reported to present as: (i) a NARP phenotype, with sensory-predominant axonal polyneuropathy; (ii) severe motor-predominant axonal polyneuropathy mimicking CMT-2; or, (iii) distal hereditary motor neuropathy (dHMN, distal axonal pure-motor neuropathy). 28 Finally, the classical, late-onset, and pure HSP described in the proband, significantly differs from previously published cases as she did not have developmental or multisystemic issues.
These cases highlight the diagnostic challenge of ATP6-related disorders and the possible mimic of “classic” HSP. Importantly, mitochondrial DNA genetic testing should be considered for unsolved HSP patients. Treatment options in ATP6-related disorders can include a mitochondrial cocktail aiming to improve OXPHOS-derived ATP production, though the evidence base for their use is generally limited. This combination of therapy was reported to subjectively improve energy and balance in two other patients with the m.9035T > C variant in the MT-ATP6 gene. 20 Of note, no significant effect was observed in the present proband so far. Further studies are required to evaluate the effect of this cocktail in these patients.
Conclusion
In summary, the phenotypic spectrum of ATP6-related disorders spans a broad group of conditions from HSP to spastic ataxia and neurodevelopmental disorders, and MT-ATP6 variants represent a very rare mitochondrial etiology of HSP. This diagnosis can be especially challenging as patients may not present with multisystemic involvement evocative of mitochondrial disorders and age-dependent or incomplete penetrance may be present. MT-ATP6 pathogenic variants may present as complex, but also pure HSP, as in the present work, without any other clinical clues for a mitochondrial origin apart from maternal transmission. Interestingly, the MT-ATP6 gene is typically not covered by most HSP panels. Clinicians and geneticists should however be aware of the small proportion of HSP caused by mitochondrial variants and consider adding mitochondrial testing to existing HSP diagnostic evaluations.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602251391068 - Supplemental material for MT-ATP6 variant as a cause of adult-onset hereditary spastic paraparesis: A case report and literature review
Supplemental material, sj-docx-1-jnd-10.1177_22143602251391068 for MT-ATP6 variant as a cause of adult-onset hereditary spastic paraparesis: A case report and literature review by Lola ER Lessard, Danielle K Bourque, Pierre J Bourque, Hanns Lochmüller, Joaquin Machado, Giulia F Del Gobbo, Aren E Marshall, Ian C Smith, Care4Rare Canada Consortium, Kym M Boycott, Jodi Warman-Chardon and Ari Breiner in Journal of Neuromuscular Diseases
Footnotes
Acknowledgments
The authors thank the patients for participating in this study.
ORCID iDs
Patient consent statement
The patients provided written informed consent for the publication of the manuscript. Approval for the Care4Rare Canada study was acquired from the Children's Hospital of Eastern Ontario Research Ethics Board (CTO-1577, 08/71X). Patients provided informed consent to be enrolled in the study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was performed under the Care4Rare Canada Consortium funded by Genome Canada and the Ontario Genomics Institute (OGI-147), the Canadian Institutes of Health Research, Ontario Research Fund, Genome Alberta, Genome British Columbia, Genome Quebec, and Children's Hospital of Eastern Ontario Foundation. LERL is supported by the postdoctoral fellowship grant from AFM-Téléthon (Grant# 28653).
Declaration of conflict of interest
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Conflict of interest: HL is an Editor-in-Chief at the Journal of Neuromuscular Diseases but had no involvement in the editorial decisions or peer review process of this manuscript. The authors declare that there are no other conflicts of interest regarding this publication.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.