
Abstract
Background:
Amyotrophic lateral sclerosis (ALS) is a severe neurodegenerative disorder that progressively affects motor neurons. Gain-of-function mutations in serine palmitoyltransferase (SPT) genes, notably SPTLC1 and SPTLC2, have been linked to juvenile ALS. Here, we describe two childhood-onset ALS cases with distinct SPTLC2 mutations, providing new insights into sphingolipid dysregulation and its role in ALS pathogenesis.
Material and methods:
Two Chinese patients with early-onset ALS, both carrying SPTLC2 mutations, were recruited from Beijing Children's Hospital. We conducted whole-exome sequencing (WES) to identify genetic variants, followed by Sanger sequencing for validation. Sphingolipid profiles were analyzed using ultra-high-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). Clinical evaluations included neurological assessments, brain MRI and electromyography. Additionally, mutant cell lines were established to assess the functional effects of the specific mutations.
Results:
Patient 1, a 6-year-old male, exhibited a novel heterozygous de-novo SPTLC2 variant (c.197T > G, p.T66R). Patient 2, a 7-year-old female, had a recurrent heterozygous de-novo SPTLC2 variant (c.778G > A, p.E260K). Both patients showed elevated levels of specific sphingolipids compared to controls, with distinct profiles between the SPTLC2-ALS and SPTLC1-hereditary sensory and autonomic neuropathy type 1 (HSAN1) cases. The novel p.T66R mutation was predicted to alter protein interactions within the SPT complex, potentially impairing sphingolipid homeostasis. Functional studies further revealed that the p.T66R variant reduces the inhibitory regulation of SPT by ORMDL proteins, leading to unrestrained SPT activity and excess sphingolipid production.
Conclusions:
Our findings identify a novel SPTLC2 variant linked to childhood-onset ALS and reveal altered sphingolipid profiles associated with different genetic mutations. These results underscore the importance of sphingolipid metabolism in ALS and suggest potential avenues for targeted therapeutic interventions. Further research is needed to explore treatment options aimed at modulating sphingolipid levels and correcting genetic defects, as well as investigating potential biomarkers for early diagnosis.
Keywords
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder characterized by the degeneration of upper and lower motor neurons. ALS exhibits significant genetic heterogeneity, with various genetic mutations contributing to its onset and progression. Recent studies have highlighted the role of gain-of-function mutations in the serine palmitoyltransferase (SPT) genes, SPTLC1 and SPTLC2, as critical contributors to childhood-onset ALS.1–10
SPT, a key enzyme in sphingolipid biosynthesis, catalyzes the rate-limiting condensation of L-serine and palmitoyl-CoA to produce 3-ketodihydrosphingosine (3-KDS). This pathway is tightly regulated to maintain cellular homeostasis, particularly in the nervous system, where sphingolipids are crucial components of myelin and neuronal membranes. Dysregulation of SPT activity due to mutations in SPTLC1 or SPTLC2 can lead to the overproduction of toxic sphingolipid species, which has been implicated in the selective vulnerability of motor neurons observed in ALS. 11
While SPTLC1 and SPTLC2 mutations have been more commonly associated with hereditary sensory and autonomic neuropathy type 1 (HSAN1), recent evidence has shown that specific mutations in SPTLC1 and SPTLC2 can also lead to childhood-onset ALS. Only 12 cases of ALS linked to SPTLC2 mutations have been reported till now, underscoring the rarity and clinical significance of these genetic variants.1–3 These mutations often manifest with severe clinical features, including progressive limb weakness, bulbar symptoms, and respiratory complications.1–3
Given the emerging role of SPT mutations in ALS, there is a growing need to understand the biochemical and molecular mechanisms underlying sphingolipid dysregulation in these patients. Recent studies suggest that these mutations lead to an imbalance in sphingolipid homeostasis, driving neurodegeneration through mechanisms that are still being elucidated. 11
In this study, we focus on two Chinese patients with childhood-onset ALS who carry distinct SPTLC2 mutations. Our aim is to elucidate the genetic and biochemical underpinnings of these cases, providing insights into how specific SPTLC2 variants contribute to disease pathology and identifying potential biomarkers for diagnosis and therapeutic targets.
Material and methods
Participants
Two Chinese patients carrying SPTLC2 mutations were recruited from the Department of Neurology, Beijing Children's Hospital, Capital Medical University between March 2024 and August 2024. The patients were evaluated by neurological examination which included the Medical Research Council (MRC) scale for assessing muscle strength. Clinical data of the patients, including age at onset, clinical manifestations, laboratory results, and genetic analysis results were collected. The study protocol ([2023]-E-106-Y) was approved by the Medical Ethics Committee of Beijing Children's Hospital, Capital Medical University. Written informed consent for routine and investigative studies was obtained from the parents of all the patients.
Whole-exome sequencing
Genomic DNA was extracted from whole blood samples of the two patients (patients 1–2) and their parents. Whole-exome sequencing (WES) of the two probands and their parents was performed as previously described. 12 To validate the mutations identified by WES, PCR amplification of the mutation site was followed by Sanger sequencing.
Analysis of sphingolipids in patient plasma samples using UPLC-MS/MS
100 μL of plasma was added to a 2 mL centrifuge tube with 900 μL methanol. The mixture was sonicated for 10 min and centrifuged at 12,000 rpm for 10 min at 4 °C. A 600 μL supernatant was transferred to a glass vial for testing. The extraction samples and standards were analyzed for sphingolipids by UPLC-MS/MS (ultra-high-performance liquid chromatography tandem mass spectrometry, TQ-S, Waters, USA) according to the previous literature with some modifications.1–4,13–15 5 μL of the samples were injected onto a Waters BEH Amide column using water (added 0.1% formic acid and 10 mM ammonium formate) (A) and isopropanol:methanol (9:1) (added 0.1% formic acid and 10 mM ammonium formate) (B) as the mobile phases for chromatographic separation. A flow of 95% B was maintained for 0.5 min, 95% to 70% B for 0.5–3 min, 70% to 50% B for 3–4 min, 50% to 95% B for 4–4.1 min, 95% was maintained for 4.1–5 min. The column was operated at 35 °C and with a flow rate of 0.25 mL/min. The mass spectrometry parameters were optimized by selecting the positive ion mode for the system. The operating conditions were as follows: capillary voltage was 3 kV, desolvation gas temperature was 500 °C, source temperature was 150 °C, cone gas flow rate was 150 L/h, desolvation gas flow rate was 800 L/h. In the patient's sphingolipid detection experiment, we employed an isotopically labeled internal standard, C16-Ceramide-d31 (CAS No.: 852043-41-5), to ensure quality control and normalization during quantification. This internal standard was added to all samples prior to lipid extraction, and peak areas were normalized accordingly. We referred to previously reported literature, 13 and the quantification was relative. The multiple reaction monitoring (MRM) method for each analysis is provided (see Table S1).
Plasmids
Plasmid for expression of human SPTLC2 cDNA by the CMV promoter was purchased from OriGene Technologies (Cat# SC127529) Inc. (Rockville, MD). The p.T66R and p.E260K variants in human SPTLC2 were introduced by QuikChange mutagenesis (Agilent Technologies) and verified by sequencing.
Antibodies
Antibodies for detecting SPTLC1 (BD Biosciences, Catalog # 611304), SPTLC2 (Protein Tech 51012-2-ap), ORMDL (EMD Millipore ABN417), GAPDH (Santa Cruz SC-47724), and Calnexin (Santa Cruz SC-11397) were used. The SPTSSA antibodies were generated by New England Peptide (now Vivetide, Gardner, MA) as described earlier. 16
Cell culture, transfections and heavy serine labeling
HEK293 cells (American Type Culture Collection, Manassas, VA) were cultured as previously described.4,17 Briefly, cells were grown in DMEM with high glucose, pyruvate, and glutamax, supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin. Lipofectamine 2000 (ThermoFisher) was used for transfections according to the manufacturer's instructions. HEK293 SPTLC2 KO cells (6.5 × 105) were plated in 6 well plates and transfected with 800 ng each of plasmids expressing WT or mutant SPTLC2. When deuterium-labeled serine (3,3-D2 L-Serine, DLM-161, Cambridge Isotope Laboratories) was used, it was added to a final concentration of 3 mM for 24 h before harvesting. Cells were harvested, washed in PBS, suspended in 50 mM Tris, pH 7.5, 1 mM EDTA, 15% glycerol with protease and phosphatase inhibitors and split to glass tubes for lipid extraction and Eppendorf tubes for protein quantification. The HEK293 SPTLC2 knockout (KO) cells were generated using a kit (KN402420) purchased from Origene. HEK293 cells were transfected with the pCAS9-Guide plasmid and linear RFP-P2A-BSD donor fragment according to the manufacturer's instructions. In brief, 3 × 105 cells were transfected and passaged seven times before applying blasticidin for selection. After isolation of colonies on 10-cm dishes, clones were subcloned to 24-well dishes. Single colonies were isolated and propagated for identification of deletion clones in 12 wells. Candidate KO lines were identified by Genomic PCR. Homozygous KO lines were verified by genomic PCR, Western blotting and by functional analysis using d2 serine labeling followed by MS.
SPT assays
Microsomes were prepared from HEK SPTLC2 KO cells transfected with WT or mutant SPTLC2 and control HEK cells as previously described. 18 Reactions were initiated by adding 50 μg of microsomal membrane to a reaction buffer (final volume of 300 μl) containing 50 mM Hepes, pH 8.1, 50 μM pyridoxal 5’-phosphate, 25 μM palmitoyl-CoA, 2.5 mM serine and 20 μCi of [3H] serine. BSA or BSA-C8-ceramide (Avanti Polar Lipids) complex (each at 0.02 mM) were prepared as described. 19 After 10 min, NH4OH was added to a final concentration of 0.25 M, followed by 2 ml of CHCl3/methanol (1:2), and the mixture was vortexed. Sphingoid bases were extracted by adding 1 ml of CHCl3 and 2 ml of 0.5 M NH4OH, vortexing and briefly centrifuging. The upper aqueous layer was removed and the lower layer was washed with 2 ml of 60 mM KCl and centrifuged. The washing was repeated twice and 1 ml of the sample was dried and counted.
Western blotting
Cells were lysed by sonication in 50 mM NaCl, 25 mM Tris-HCl pH 7.5, 1 mM EGTA, 10% glycerol, 0.3% NP-40, 0.3% deoxycholate and 0.03% SDS with protease and phosphatase inhibitors (complete protease inhibitor and PhosSTOPphosphatase cocktails, Sigma Aldrich). Protein concentrations were determined by Bio-Rad assay. Proteins were diluted into 1 × LDS sample buffer with reducing agent (Thermofisher), resolved by electrophoresis on NuPAGE Novex 4–12% Bis-Tris Gels using MES buffer (Thermofisher) and transferred to a nitrocellulose membrane. Membranes were blocked in Intercept blocking buffer (LI-COR) in TBS for 1 h, incubated with primary antibody in the same buffer in TBST, washed and incubated with secondary antibody, IRDye 800CW goat anti-rabbit or goat anti-mouse (LI-COR Biosciences). The bands were visualized using the Odyssey system (Odyssey CLx) and Image Studio Lite version 5.5.
Cellular sphingolipid analysis using HPLC and mass spectrometry
Cell pellets (0.1–0.2 mg) were added to 1 ml methanol containing 25 pmol of internal standards (Avanti Polar Lipids, LM6002) and bath sonicated for several minutes. Following addition of 0.5 ml chloroform, samples were incubated overnight at 48 °C, briefly centrifuged to remove insoluble material and dried under nitrogen. The HPLC mobile phase solvents were (A) CH3OH:H2O:CH2O2 (74:25:1, v/v/v with 5 mM Ammonium Formate) and (B) CH3OH:CH2O2 (99:1, v/v with 5 mM Ammonium Formate). The dried samples were resuspended in 0.2 ml of solvents A and B at 80:20 (v/v), bath sonicated, centrifuged to clarify and transferred to an HPLC vial for analysis. Typically, 5–20 μl of sample were injected. The samples were analyzed using an Agilent 1200 Series HPLC coupled to an ABSciex QTRAP 4000 MS. The column (75 mm × 4.6 mm × 5 μm, Supelco Discovery BIO Wide Pore C18 HPLC) was pre-equilibrated with A–B (80:20 v/v) and held for 2 min after injection at 0.5 ml/min. The flow was ramped to 100% B at 7 min, held for 30 min, and quickly ramped back to 80:20 and held for 2 min before the next injection. The mass spectrometer was set to detect compounds in MRM mode in a dual period run with long-chain bases detected in each run (from 3–9.2 min) and each class of sphingolipid (Cer, hexosylCer, SM) detected in separate runs (from 9.2 to 30 min). The MRM parameters were as described. 20 Compounds were quantified by taking the ratio of the peak area to the known concentration of the representative internal standard using the ABSciex Analyst program and normalized to protein concentration. Because of the MRMs used, the contribution of naturally occurring isotopes to the +2 species being quantitated was negligible except in the case of sphingomyelins, which were corrected using a commercially available isotope distribution calculator (www.sisweb.com).
Results
Phenotype of patients
Patient 1 is a 6-year-7-month-old male presenting with motor developmental delay since infancy, followed by regression, characterized by worsening weakness in limbs. By the age of 3 and a half years, the patient was unable to walk with support, and by 4 years old, he could not sit independently. At 4 years and 6 months, scoliosis was noted, which has since shown progressive worsening. The patient demonstrates adequate comprehension, calculation ability, and memory, but experiences difficulties with speech, including choking while drinking water and swallowing. There is no perinatal medical or family history reported. Developmentally, the patient achieved head lifting at 5 months, independent sitting at 12 months, and walking with support at 18 months, but has never achieved independent walking. Speech development has been delayed, with the patient starting to say simple words at 2 years old and simple sentences at 3 and a half years old. The physical examination revealed muscle atrophy throughout the body, tongue atrophy (Figure 1A) with fasciculations, and gaze-evoked nystagmus during lateral gaze. Muscle strength was decreased in both upper and lower limbs, graded as 3 proximally and 3 + distally in the upper limbs, and grade 2 proximally and grade 3 distally in the lower limbs. The biceps and triceps brachii tendon reflexes, as well as the knee reflexes, were absent bilaterally, while the Achilles tendon reflexes were hyperactive bilaterally. Additionally, bilateral Babinski sign was positive, and bilateral ankle clonus was positive. Auxiliary examinations included a normal CK level, EEG and brain MRI. Nerve conduction studies showed normal motor conduction velocities, but the CMAP (compound muscle action potential) amplitudes of the bilateral common peroneal nerves were reduced. Sensory conduction velocities and amplitudes were normal. Electromyography demonstrated prolonged duration and increased amplitude of the MUAP (motor unit action potential), and revealed chronic neurogenic damage.
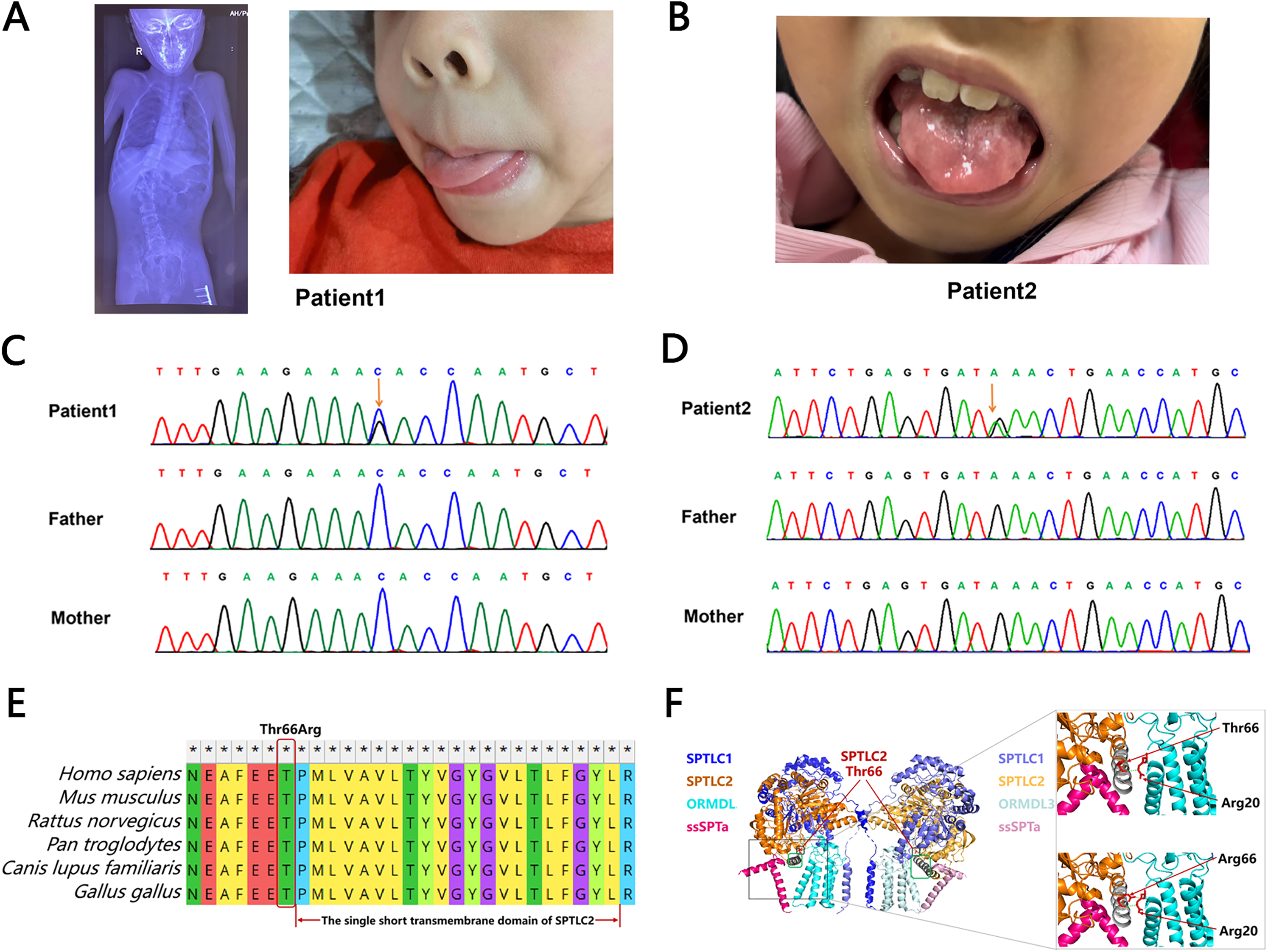
Genotype, phenotype, and structural effects of SPTLC2 p.T66R. (A) Clinical pictures showing X ray of spine scoliosis and tongue atrophy in the patient 1. (B) Clinical pictures showing scalloped tongue in the patient 2. (C) Sanger sequencing illustrating the de novo c.197T > G variant of the SPTLC2 in the patient 1's family. (D) Sanger sequencing illustrating the de novo c.778G > A variant of the SPTLC2 in the patient 2's family. (E) T66R is highly evolutionarily conserved across different species. (F) The tertiary model of the SPT complex includes SPTLC1 (blue-purple), SPTLC2 (yellow), ORMDL3 (blue-green), and SSPTa (pink). Zooming in reveals the interaction between ORMDL3 (blue-green) and the 66th amino acid of SPTLC2.
Patient 2 is a 7-year-9-month-old female presenting with a motor developmental delay and progressive limb weakness lasting over 4 years. At 3.5 years old, she displayed weakness in both lower limbs, leading to gait abnormalities and the need for rest after walking a few dozen steps. By 4 years and 9 months, she developed dysphonia, characterized by unclear articulation and difficulty swallowing liquids without choking. At 5 years old, she could no longer walk independently, and by 7 years and 2 months, she began showing instability while sitting alone. Her limb weakness has progressively worsened, and she currently cannot sit independently. Despite these issues, her intellectual abilities, including calculation, comprehension, and memory, remain normal. There is no reported perinatal medical or family history of note. Her intellectual and physical development milestones align with those of her peers: she held her head up at 2 months, sat up at 7 months, walked at 12 months, and spoke simple words by 18 months. Physical examination revealed generalized muscle wasting, including the muscles of the face. She could track visually, smile, and understand, but had weak neck muscles, a low voice, and atrophied tongue muscles with fasciculations (Figure 1B). Muscle tone was reduced in all four limbs, with significant proximal weakness: proximal muscle strength was 3 in the upper limbs and 2 in the lower limbs, while distal muscle strength was 4 in the upper limbs and 3 in the lower limbs. Knee and ankle joint contractures were present, and neither knee nor tendon reflexes were elicited; pathological reflexes were negative. However, at the age of 4 years and 6 months, the physical examination showed increased muscle tone, bilateral ankle clonus was positive, knee tendon reflexes were hyperactive, and Babinski sign was positive. Auxiliary examinations showed normal CK levels, EEG, and brain MRI. Nerve conduction studies showed no significant characteristic changes in the examined sensory nerves, while the CMAP amplitudes of the examined motor nerves were significantly reduced. Electromyography revealed that the MUAPs of the examined muscles had prolonged durations and significantly increased amplitudes, indicating chronic neurogenic damage.
Genotype of patients
Genomic DNA was extracted from the affected probands and their unaffected biological parents, and trio-based whole-exome sequencing (trio-WES) was performed. In patient 1, we identified a novel heterozygous de novo variant in SPTLC2 (NM_004863: c.197T > G, p.T66R), which has not been previously reported. In patient 2, a recurrent heterozygous de novo variant in SPTLC2 (NM_004863: c.778G > A, p.E260 K), previously associated with ALS,1–3 was detected. Both variants were subsequently validated by Sanger sequencing (Figures 1C and 1D). Combined analysis of WES and Sanger results confirmed that the variants were absent in the parents, thus verifying their de novo origin. These findings support the potential pathogenicity of the identified SPTLC2 mutations.
The novel variant p.T66R substitutes a conserved threonine residue with arginine (Figure 1E). Based on the tertiary model of the SPT complex,21,22 we predicted structural changes between the wild type and the SPTLC2 p.T66R mutation (Figure 1F). In the wild type, the 66th amino acid threonine of the SPTLC2 protein forms a hydrogen bond with the 20th amino acid arginine of the ORMDL3 protein. In contrast, for the mutant type, the 66th amino acid arginine of the SPTLC2 protein forms two hydrogen bonds with the 20th amino acid arginine of the ORMDL3 protein. These changes in hydrogen bonding at these positions may alter the interaction between SPT and ORMDL3, leading to impaired inhibition of SPT by ORMDL3 and disrupting sphingolipid homeostasis. Therefore, our trio whole-exome and tertiary model analysis strongly suggested that the novel missense variant (c.197T > G, p.T66R) in SPTLC2 was a causative factor in early-onset ALS.
Sphingolipids profiles of patients
To verify the pathogenicity of the mutations and analyze sphingolipid levels, plasma samples were collected from the SPTLC2-related ALS and SPTLC1-related HSAN patients, as well as from seven healthy controls, including unaffected parents of patients SPTLC2-ALS1 and SPTLC2-ALS2, and three additional healthy pediatric controls. The HSAN patient had a mutation in the SPTLC1 gene, c.1015 G > A (exon 11), p.Ala339Thr, which had been previously reported to be associated with HSAN. 23 Sphingolipid levels were analyzed using ultra-high-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS), revealing distinct sphingolipid profiles between the patients and controls.
As shown in Figure 2, significant differences in sphingolipid species were observed among the Normal, HSAN, and ALS groups. The levels of 1-deoxysphingolipids, specifically Cer(m18:0/16:0), Cer(m18:0/20:0), and Cer(m18:0/22:0), were markedly elevated in the HSAN patient compared to both the normal controls and ALS patients (Figures 2A-2C). In contrast, these metabolites remained low and comparable between the ALS patients and healthy individuals, suggesting HSAN-specific deoxysphingolipid accumulation.

Plasma sphingolipid profiles in SPTLC2-ALS patients, SPTLC1-HSAN patients, and normal controls. (A-C) Levels of Cer(m18:0/16:0), Cer(m18:0/20:0), and Cer(m18:0/22:0) were significantly elevated in HSAN patients compared to both normal controls and ALS-P1 and ALS-P2 patients. (D-E) Levels of Cer(d18:0/16:0) and SM(18:1/18:1) were significantly increased in ALS-P1 and ALS-P2 patients compared to both normal controls and HSAN patients. (F) Levels of SM(d18:0/16:0) were elevated in both ALS and HSAN patients compared to normal controls. For bar graphs, data are presented as mean ± SD. Comparisons were performed using the T-test. ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. There are seven normal controls, including unaffected parents of patients SPTLC2-ALS1 and SPTLC2-ALS2, and three additional healthy pediatric controls.
Compared with the accumulation pattern of deoxysphingolipids, certain canonical sphingolipids exhibited a more pronounced elevation in ALS patients. For example, Cer(d18:0/16:0) and SM(d18:1/18:1) were significantly increased in ALS patients compared to both healthy controls and the HSAN patient (Figure 2D-2E). In addition, other canonical sphingolipids were elevated in both ALS and HSAN patients, such as SM(d18:0/16:0) (Figure 2F), suggesting that these metabolic changes may reflect broader perturbations of the SPT-related pathway.
In summary, these results reveal disease-specific alterations in sphingolipid metabolism. The HSAN patient exhibited a selective elevation of deoxysphingolipids and certain canonical sphingolipids, whereas ALS patients were characterized by the accumulation of canonical ceramides and sphingomyelins. These findings support the notion that different mutations in the SPTLC1 or SPTLC2 genes may lead to distinct, disease-specific patterns of sphingolipid dysregulation, suggesting divergent pathogenic mechanisms associated with each variant.
The effects of SPTLC2 variants on SPT activity and ORMDL regulation
To directly investigate how the SPTLC2 variants affect SPT activity, de novo SPT activity was investigated. Empty vector, or plasmids expressing the WT, T66R or E260 K SPTLC2 variants were transfected into HEK SPTLC2 KO cells and after 24 h. d2-serine was added to the media for 24 h. Cells were harvested, expression of SPTLC2, SPTLC1 and ORMDL3 was assessed by immunoblotting (Figure 3A) and newly synthesized d2-serine labeled sphingolipids were analyzed by LCMS (Figure 3B). The results showed significantly higher levels of d2-serine labelled sphingolipids in cells expressing SPTLC2T66R and SPTLC2E260K compared to cells expressing WT SPTLC2. These results are consistent with the elevated sphingolipids detected in patient plasma samples.
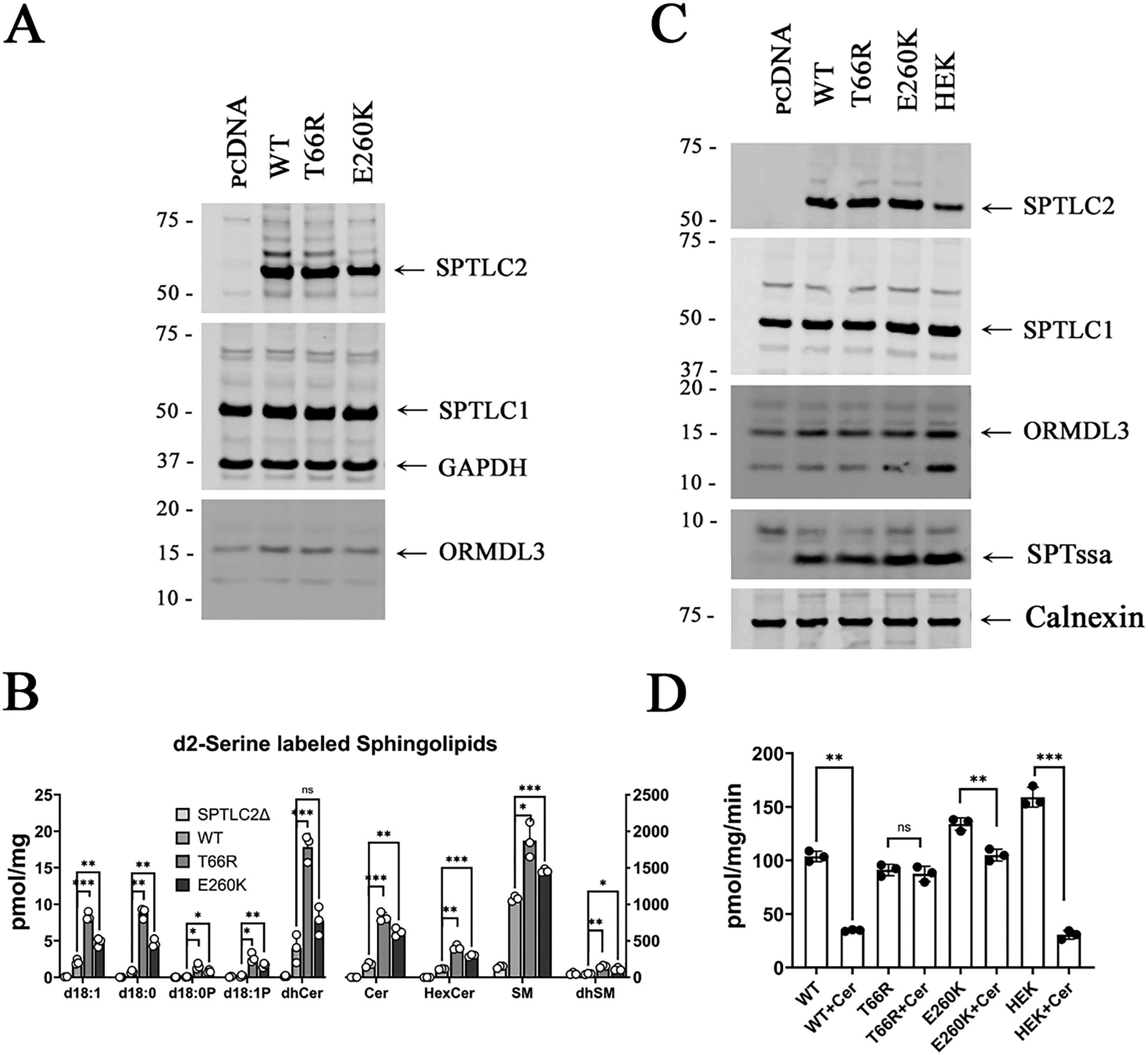
The T66R and E260K variants in SPTLC2 impair ORMDL regulation of SPT. (A) Proteins from HEK SPTLC2 KO cells expressing the WT and SPTLC2 variants were separated by SDS-PAGE and the indicated proteins were analyzed by immunoblotting. (B) Following incubation with d2-serine, sphingolipids were extracted from the cells (as in A) and analyzed by LCMS. Statistical significance was determined using a 2-tailed t-test with Welch's correction in GraphPad Prism; (C) Microsomal proteins from SPTLC2 KO cells transfected with pcDNA, the SPTLC2 variants, or from control HEK cells were separated by SDS-PAGE and immunoblotted for the indicated proteins. (D) Microsomes (as in C) were assayed for SPT activity with or without added C6-ceramide as described in the Materials and Methods.
Several recent studies of variants in the subunits of SPT from patients with neurodegenerative disease, including the SPTLC2E260K variant, have revealed that these mutations in SPT interfere with the negative feedback regulation of SPT by the ORMDL proteins. Moreover, it has been shown that inhibition of SPT by the ORMDLs results from a ceramide-induced conformational change that locks the N-terminus of the ORMDL into the active site of SPT 24 and that the ceramide-induced inhibition of SPT can be detected using microsomal SPT assays. 19 Thus, to investigate whether the elevated SPT activity associated with the SPTLC2T66R variant similarly reflects loss of ORMDL regulation, microsomal SPT was measured in the absence and presence of C6-ceramide. As expected, SPT activity in microsomes prepared from control HEK cells and from SPTLC2 KO cells expressing WT SPTLC2 is significantly reduced by the addition of C6 ceramide (Figure 3D). In contrast, the effect of ceramide on SPT activity is reduced or abolished with SPTLC2E260K or SPTLC2T66R respectively (Figure 3D). These results conclusively show that the SPTLC2T66R mutation significantly ablates ceramide-mediated ORMDL inhibition of SPT. The growing number of disease-causing variants in the subunits of SPT that impair ORMDL regulation and result in unrestrained SPT activity point to the key role ORMDL-mediated regulation of sphingolipid homeostasis plays in the nervous system.
Discussion
In this study, we identified two de novo heterozygous mutations in SPTLC2—one novel (p.T66R) and one previously reported (p.E260K)—in two unrelated patients with childhood-onset ALS. These findings expand the mutational spectrum of SPTLC2-related ALS and highlight the critical role of sphingolipid dysregulation in its pathogenesis. Through integrative genetic, biochemical, and functional analyses, we demonstrate that these mutations disrupt sphingolipid homeostasis by impairing the ORMDL protein-mediated negative feedback regulation of SPT.
Clinically, both patients presented with early motor developmental delay, progressive weakness, and upper and lower motor neuron signs, consistent with childhood-onset ALS. Electrophysiological examinations revealed chronic neurogenic damage, and whole-exome sequencing confirmed the de novo origin of both variants. Structural modeling suggested that the novel p.T66R mutation disrupts a critical hydrogen bond between SPTLC2 and ORMDL3, similar to the known p.E260K mutation. These alterations likely destabilize the SPT-ORMDL regulatory complex and contribute to elevated SPT activity.1,2
Biochemical profiling using ultra-high-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) revealed distinct differences in sphingolipid metabolism between ALS and HSAN patients. In the two ALS cases, significant accumulation of canonical sphingolipids was observed, including Cer(d18:0/16:0) and SM(d18:1/18:1) (Figure 2D-E), consistent with previous reports indicating that ALS-associated mutations impair ORMDL-mediated negative feedback regulation of SPT, resulting in increased enzymatic activity and upregulated flux through the canonical sphingolipid biosynthetic pathway.4,6,11
Notably, in the HSAN patient harboring the SPTLC1 p.Ala339Thr mutation, substantial elevations were detected in 1-deoxysphingolipids such as Cer(m18:0/16:0), Cer(m18:0/20:0), and Cer(m18:0/22:0). There were also elevated levels of some of the canonical species, such as SM(d18:0/16:0) (Figure 2F). This is consistent with an earlier report 23 and studies have shown that the S331F mutation in SPTLC1 exerts dual pathogenic effects, simultaneously weakening ORMDL-mediated SPT inhibition and altering substrate specificity, thereby promoting the co-accumulation of neurotoxic deoxysphingolipids and selected canonical sphingolipids. 25 However, whether this is the case for the SPTLC1 A339T variant requires further studies of its effects on SPT activity and validation in larger cohorts.
Further mechanistic insight into the SPTLC2 variants reported herein was provided by in vitro functional assays using HEK293 SPTLC2 knockout cells. Both T66R and E260K variants resulted in increased de novo sphingolipid synthesis, as evidenced by elevated d2-serine-labeled products. Importantly, microsomal SPT assays revealed that ceramide-induced inhibition of SPT was intact in wild-type cells but significantly reduced or abolished in cells expressing SPTLC2-E260K and SPTLC2-T66R, respectively. These results demonstrate that the T66R variant, like E260K, impairs ceramide-mediated negative regulation of SPT by ORMDL proteins, leading to unrestrained enzyme activity.
Taken together, our data support a unifying model in which pathogenic SPTLC2 mutations disrupt ORMDL-mediated regulation of SPT, resulting in excessive sphingolipid biosynthesis and altered lipid composition in neuronal tissues.1–3 Notably, the specific lipid profile and clinical severity may depend on the nature of the mutation and its impact on enzyme function and feedback sensitivity. These findings underscore the importance of sphingolipid homeostasis in motor neuron integrity and point to the SPT-ORMDL axis as a potential therapeutic target. 6
Our findings emphasize the need for targeted therapeutic strategies to correct sphingolipid dysregulation in SPTLC2-related ALS. Modulating SPT activity pharmacologically, for example through agents like myriocin, has shown potential in preclinical models, though concerns regarding toxicity limit its clinical use. 26 Recent advances in transcript-targeted therapies suggest that allele-specific RNA interference, such as siRNAs selectively silencing mutant SPTLC1 alleles, may offer a promising strategy to restore sphingolipid homeostasis in SPT-related disorders. Although further optimization is needed for effective delivery to motor neurons or glial cells, such precision approaches could be explored in clinical trials for severe pediatric neurodegenerative diseases. Similar RNA-based strategies or genome-editing technologies may potentially be extended to SPTLC2-associated ALS, providing a rationale for the development of gene-targeted therapies. 4
While current treatment for ALS remains largely supportive, including neuroprotective agents, physical therapy, and respiratory management, the development of disease-modifying therapies that address underlying metabolic derangements is crucial. Notably, while serine supplementation has been explored as a potential therapeutic strategy for HSAN, where it can help mitigate the production of neurotoxic deoxysphingolipids, this approach is not suitable for ALS patients with SPTLC2 mutations. In ALS, serine supplementation could exacerbate the dysregulation of sphingolipid metabolism, potentially worsening the disease. Thus, precise understanding of the lipidomic consequences of different SPTLC mutations is essential for guiding personalized therapeutic strategies.
In summary, our study identifies a novel de novo SPTLC2 variant and confirms a recurrent pathogenic mutation, both associated with childhood-onset ALS. These findings expand the genetic and biochemical landscape of SPTLC2-related ALS and underscore the central role of sphingolipid dysregulation in its pathogenesis. By combining genetic, biochemical, and functional evidence, we demonstrate that disrupted ORMDL-mediated regulation of SPT leads to excessive sphingolipid production, which may contribute to motor neuron degeneration. These insights not only enhance our understanding of disease mechanisms but also suggest potential therapeutic avenues targeting sphingolipid metabolism. Further research is warranted to validate these findings in larger cohorts and to explore targeted interventions for this rare but severe form of motor neuron disease.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602251370586 - Supplemental material for Characterization of novel and recurrent SPTLC2 variants in childhood-onset amyotrophic lateral sclerosis: Insights into sphingolipid dysregulation
Supplemental material, sj-docx-1-jnd-10.1177_22143602251370586 for Characterization of novel and recurrent SPTLC2 variants in childhood-onset amyotrophic lateral sclerosis: Insights into sphingolipid dysregulation by Xiaona Fu, Kenneth Gable, Sita D. Gupta, KaiLi Zhang, Bingbing Jia, Wenjun Wang, Xinying Yang, Lu Wang, Lin Ge, Carsten G Bönnemann, Teresa M Dunn and Hui Xiong in Journal of Neuromuscular Diseases
Supplemental Material
sj-xlsx-2-jnd-10.1177_22143602251370586 - Supplemental material for Characterization of novel and recurrent SPTLC2 variants in childhood-onset amyotrophic lateral sclerosis: Insights into sphingolipid dysregulation
Supplemental material, sj-xlsx-2-jnd-10.1177_22143602251370586 for Characterization of novel and recurrent SPTLC2 variants in childhood-onset amyotrophic lateral sclerosis: Insights into sphingolipid dysregulation by Xiaona Fu, Kenneth Gable, Sita D. Gupta, KaiLi Zhang, Bingbing Jia, Wenjun Wang, Xinying Yang, Lu Wang, Lin Ge, Carsten G Bönnemann, Teresa M Dunn and Hui Xiong in Journal of Neuromuscular Diseases
Supplemental Material
Footnotes
Acknowledgments
We thank the patients and their families for their participation in this study. This work was supported by National Key R&D Program of China (No. 2022YFC2703100), National Natural Science Foundation of China (No. 82171393 and No. 82471430), Joint Basic-Clinical Laboratory of Pediatric Epilepsy and Cognitive Developmental (No. 3-1-013-03), Beijing Municipal Science & Technology Commission Proof of Concept Center (No. 2024030119), and by Award #HT94252310375 from the CDMRP Amyotrophic Lateral Sclerosis Research Program, Therapeutic Development Award (TMD). The opinions and assertions expressed herein are those of the author(s) and do not reflect the official policy or position of the Uniformed Services University of the Health Sciences or the Department of Defense.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.