
Abstract
Introduction
MLASA (myopathy, lactic acidosis and sideroblastic anemia) is a rare, autosomal recessive mitochondrial disorder. Symptom onset typically occurs in childhood and differs considerably in disease severity. Congenital-onset disease is uncommon.
Methods
Whole genome sequencing was performed which identified two heterozygous, rare YARS2 (NM_001040436.3) variants in trans, one of which was novel. The diagnosis was confirmed with muscle biopsy and mitochondrial enzyme activity testing. To compare the clinical phenotype of our patient to those previously described in the literature, we reviewed all YARS2-related cases in literature to identify age of symptom onset and associated clinical features.
Results
The maternally-inherited variant, c.948G > T, p.(Arg316Ser), was previously reported with MLASA; the paternally-inherited variant, c.917T > C, p.(Phe306Ser) has not been previously reported. Muscle biopsy showed non-specific changes, that can be seen with mitochondrial dysfunction. Mitochondrial enzyme activity testing on frozen muscle tissue confirmed reduced complex I, III and IV activities.
Discussion
In our case report we describe a patient with MLASA, caused by compound heterozygous variants in YARS2. Our child with congenital-onset disease remains stable at 23 months old. Her stable course differs from two other children with congenital-onset disease who died in the first few days to months after birth. Mitochondrial enzyme activity testing is important to establish pathogenicity of novel variants in patients with this rare and clinically heterogeneous disease.
Introduction
MLASA (myopathy, lactic acidosis and sideroblastic anemia) is a rare autosomal recessive disorder, which affects oxidative phosphorylation and iron metabolism mainly in skeletal muscle and bone marrow. It was first described in 2004 by Bykhovskaya et al., 1 although the clinical features were summarized earlier by different groups.2,3 MLASA typically manifests in childhood, presenting with a myopathy phenotype that ranges in severity, as well as lactate acidosis and sideroblastic anemia, which may be transfusion dependent. There is no curative treatment available. Three genes have been associated with similar MLASA phenotypes. MLASA1 is attributable to biallelic variants in PUS1. 4 MLASA2 is attributable to YARS2, variants and reported in 38 patients.5–17 MLASA3 has been reported in a single case associated with a missense variant in the mitochondrial gene, MT-ATP6, which gave rise to a severe phenotype. 18 We describe a patient with congenital-onset disease and biallelic variants in YARS2.
Methods
Literature search
We completed a PubMed Search for all articles containing the key words “MLASA” and “YARS2”. The reference section of each article was reviewed to identify additional articles. All English articles were included.
Genetics
Trio whole genome sequencing (WGS) was performed on clinical testing through the Ontario Genome-Wide Sequencing laboratory using TruSeq PCR-free library preparation followed by 2 × 150 bp paired and sequencing on the Illumina Novaseq6000 platform to an average depth of coverage of at least 35X. Genome alignment (Build GRCh38/hg38) and small sequence variant (substitutions, insertions, duplications and deletions) and copy number variant analysis was performed with the Illumina Dragen Germline analysis platform.
Muscle biopsy
To further investigate our findings from the genetic testing, we performed a muscle biopsy from the left vastus lateralis muscle. Routine histochemical staining and ultrastructural studies were performed.
Snap-frozen muscle sample was sent for mitochondrial oxidative phosphorylase enzyme activity (SickKids Hospital, ON). After generating homogenates from the frozen tissue, enzymes were measured using dual-beam, temperature-controlled spectrophotometry.
Case report
A 23-month- old girl first presented to our Neurology clinic at the age of 7 months due to congenital-onset ptosis and ophthalmoplegia. She was the first child to healthy, unrelated, white parents. Family history was negative for any neurological disorders. Pregnancy was unremarkable and fetal movements were normal. She was delivered by an induced vaginal birth at 37 weeks due to pre-eclampsia. Birth weight was 2.260 kg (<1st percentile). She remained in hospital for five days due to difficulty establishing feeds. Bilateral ptosis was present at birth but became more evident at one month old, at which time she was referred to Ophthalmology. Neurological examination at 7 months noted decreased length 60 cm (<1st percentile, −2.8 SD), weight 4.635 kg (<1st percentile, −4.4 SD) and head circumference 40 cm (<1st percentile, −2.7 SD). Bilateral ptosis was evident, to the level of the mid-pupil and decreased facial expression was apparent. Her palate was high arched. Appendicular tone was decreased. Muscle strength noted antigravity movements to her arms and legs with normal, 2 + deep tendon reflexes. Gross motor delay was apparent as she could only roll back-to-side but not back-to-front. She could not sit independently nor grasp her feet with her hands. Her initial hemoglobin was normal 105 g/L (range 100–125 g/L) and has remained slightly low to within normal range 96 g/L to 107 g/L, without need for pRBC transfusion. Her serum lactate has been persistently elevated, between 2.8 and 5.9 mmol/L (normal: 0.5–2.2 mmol/L). Her serum creatine kinase, acylcarnitine profile and plasma amino acids were reassuring. Echocardiogram at 11 months old showed concentric left ventricular hypertrophy with normal biventricular function and a LV ejection fraction of 64.7%. She was started on coenzyme Q10, vitamin B50, L-carnitine and creatine monohydrate. Repeat echocardiogram 6 months later demonstrated an improvement in LV function, now within normal limits for age. She underwent ptosis correction surgery at the age of 13 months. There was improvement observed following the bilateral upper eyelid ptosis repair via frontalis silicone suspension, as the eyelids were elevated so as to no longer obscure her pupils, however partial ptosis was still visible. At her last follow-up visit at 23 months old, she was able to stand and cruise along furniture. She continues to take all calories by mouth and has increased her weight to the 3rd percentile for age.
Results
WGS in our patient showed heterozygous rare variants in the YARS2 (NM_001040436.3) gene. The first variant c.948G > T, p.(Arg316Ser) in exon 3 was maternally inherited and in trans with the second variant c.917T > C, p.(Phe306Ser), in exon 2, which was paternally inherited.
The first variant was previously reported in Rudaks et al. in compound heterozygous state along with c.98C > A, p.(Ser33Ter) in exon 1. 16 The second variant observed in our patient showed an allele frequency of 0.0006% in population controls of the Genome Aggregation Database (gnomAD,GRCh38/hg38). It is a missense variant that replaces the phenylalanine with a serine at codon 306 and is predicted to impact protein function by in silico programs. This variant has not been previously described in the literature.
The muscle biopsy (Figure 1) showed non-specific changes with hypotrophy of type 1 and type 2 fibers and a moderate range of myofiber size variation with many of the fibers being smaller than expected for age with overall, mild type 1 fiber predominance. Occasional fibers demonstrated mildly increased subsarcolemmal staining on H&E suggestive of abnormal mitochondrial aggregates, but not definitive for “ragged red fibers”. Oil Red O and Sudan black stains (not shown) showed occasional fibers with mildly increased coarse lipid droplets, a common secondary feature of mitochondrial myopathies. On electron microscopy, scattered fibers with subsarcolemmal aggregates of mitochondria were found, some of which were enlarged with prominent cristae, suggestive of mitochondrial dysfunction.
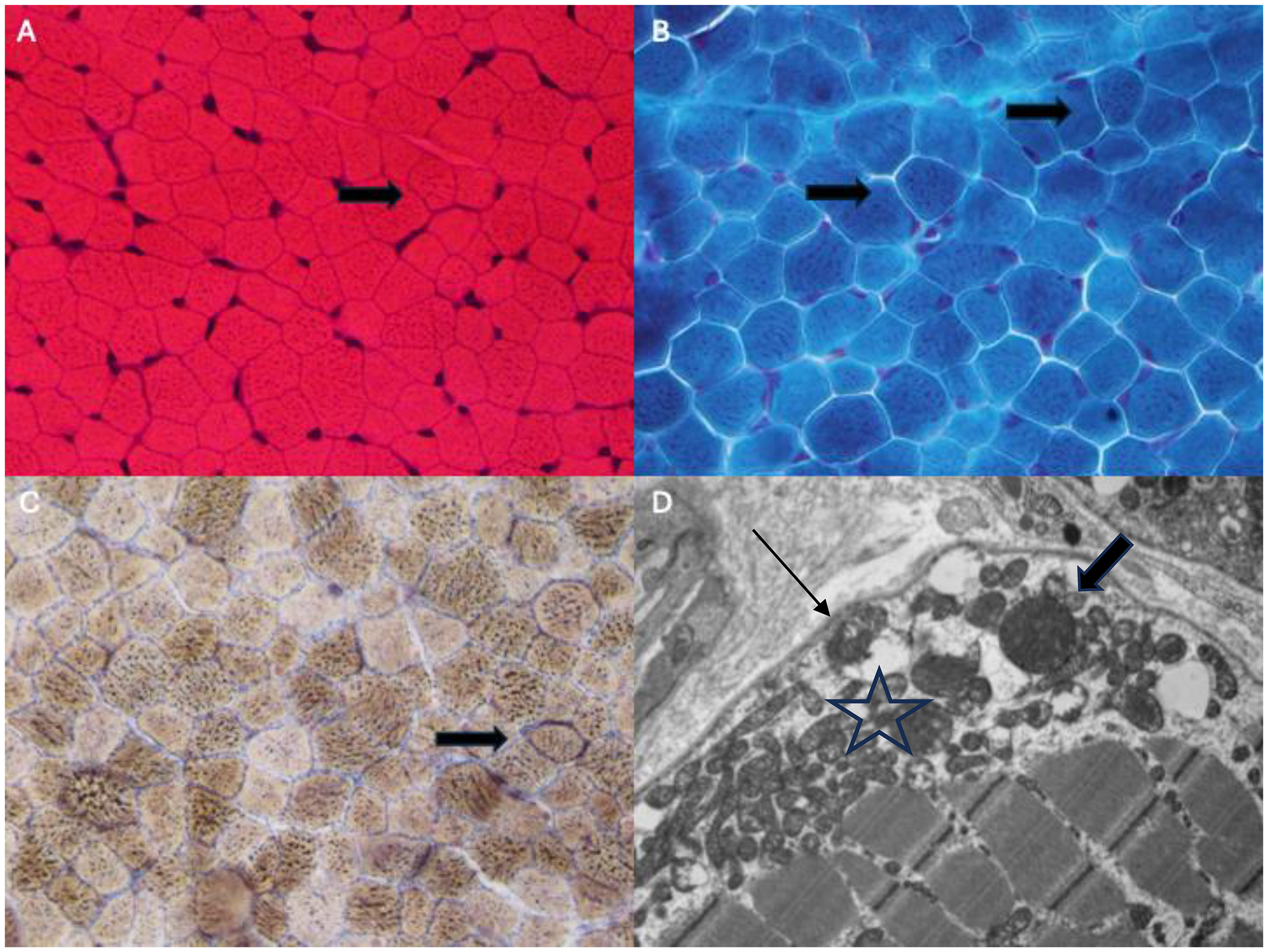
Biopsy (at 13 months old) of vastus lateralis muscle, frozen tissue. (A) H&E stain, 60X: Moderate myofiber size variation with scattered hypotrophic fibers (both type 1 and 2, not shown) and occasional fibers with increased subsarcolemmal staining (arrow). (B) Gomori trichrome stain, 60×: No definitive ragged red fibers but occasional fibers with increased coarser granular staining of their sarcoplasm (arrows). (C) Combined cytochrome C oxidase (COX) and Succinate dehydrogenase (SDH) stain, 60×: Rare fibers with increased subsarcolemmal staining suggestive of mitochondrial aggregation (arrow). No COX-negative fibers seen. (D) Electron microscopy, 9000×: A myofiber with a subsarcolemmal aggregate of mitochondria (star), some of which are enlarged with prominent cristae (wide arrow)
Mitochondrial function was assessed by respiratory chain enzyme activity assay using dual-beam, temperature-controlled spectrophotometry on homogenates from frozen tissues since isolating mitochondria from frozen tissue is not possible. The results show reduced complex IV activity (COX; complex IV 0.67 μmol/min/g wet weight; reference range 0.74–3.91) and reduced ratios of complex I + III (complex I + III/citrate synthase ratio 0.04 – reference range ≥0.10; complex I + III/complex II + III ratio 0.94 -reference range ≥0.20) These results suggest a specific defect in complex IV and is consistent with abnormal oxidative phosphorylation (OxPHOS).
The clinical presentation of our patient, the genetic findings and the results from muscle biopsy and impaired OxPHOS enzyme activity testing strongly suggests the diagnosis of MLASA, caused by compound heterozygous rare variants in YARS2.
Discussion
We describe a 23-month-old girl, with the clinical features of congenital onset MLASA, with accentuated ptosis, lactic acidosis and mild anemia. Blood transfusions have not been required. Genome sequencing found two variants of uncertain clinical significance in YARS2, one of which has not been previously reported. Together with the findings from the muscle biopsy and muscle oxidative phosphorylation (OxPHOS) enzymology, we provided evidence of pathogenicity for the variants.
Most individuals with MLASA2 develop initial symptoms during infancy or childhood (77%; 28/36) although disease-onset in adulthood has also been reported (19%; 7/36) (Figure 2).

Distribution of age of onset and frequencies of the main clinical features of MLASA2, n = 36, asymptomatic patients excluded (n = 2).
Despite the heterogeneity with regards to age of onset, clinical symptoms are common in MLASA2 with most patients showing: myopathy (72%; 26/36), lactic acidosis (72%; 26/36) or anemia (86%; 31/36) the latter often being transfusion-dependent (56%; 20/36); (Figure 2; Supplemental Table 1)
Of 38 patients have been reported with YARS2-related MLASA2, two apparently asymptomatic patients have been identified because of family history (Table 1). One of the patients has remained asymptomatic after almost 4 years of follow up and another was asymptomatic at 49 years old. 17 The asymptomatic adult patient raises the possibility of incomplete penetrance. Congenital-onset disease has only been reported in two other newborns who died at 2 days old and 3 months old. 7 While our patient does show symptoms of a mild myopathy and persistent lactic acidosis, she remains stable at 23 months with no anemia and no progressive cardiomyopathy, indicating that not all infants who show congenital onset are predicted to die in months after birth. The two other patients, siblings, with congenital onset of disease died at an early age, the first at 3 months old and the other at two days of life due to cardiopulmonary arrest. 7
Demographic, genetic and biopsy findings in patients with MLASA2.
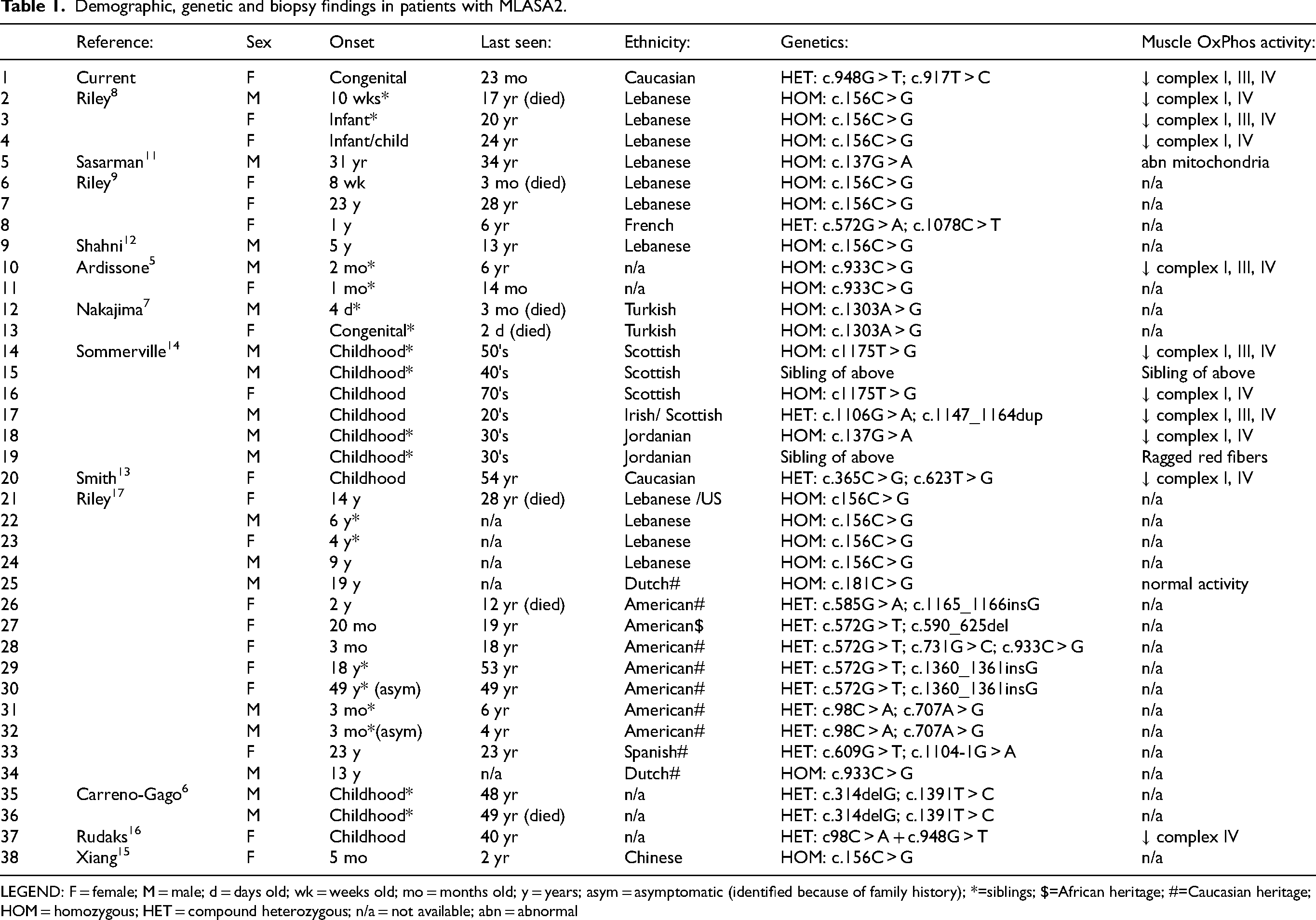
LEGEND: F = female; M = male; d = days old; wk = weeks old; mo = months old; y = years; asym = asymptomatic (identified because of family history); *=siblings; $=African heritage; #=Caucasian heritage; HOM = homozygous; HET = compound heterozygous; n/a = not available; abn = abnormal
A small proportion of symptomatic patients were reported to have died (18%; 7/36) with most (70%; 5/7) having died in childhood (< 18 yo) typically due to acute respiratory failure.6–9,17 One patient developed hemophagocytic lymphohistiocytosis, followed by hypoxic respiratory failure that led to multiorgan dysfunction, fungal sepsis and ultimately death at 28 years old. 17
Our patient demonstrates an uncommon congenital-onset phenotype with bilateral ptosis, ophthalmoplegia and failure-to-thrive being the most prominent features. Our patient, with congenital onset remains stable at 23 months old. Two other children with congenital-onset symptoms died at 2 days and 3 months old. Rapid access to WGS was very helpful at obtaining a definitive diagnosis. Although the addition of antioxidant medications coenzyme Q10, vitamin B50, L-carnitine and creatine monohydrate was associated with an improvement in cardiac function it is unclear if this was causative.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602251369227 - Supplemental material for Congenital-onset MLASA2 from a novel YARS2 variant: A literature review
Supplemental material, sj-docx-1-jnd-10.1177_22143602251369227 for Congenital-onset MLASA2 from a novel YARS2 variant: A literature review by Astrid Eisenkölbl, Hanns Lochmüller, Melissa T Carter, Leslie E Hamilton and Hugh J McMillan in Journal of Neuromuscular Diseases
Footnotes
Consent for publication
All authors carefully read the manuscript and approved it for publication.
Authors’contribution
All authors listed reviewed the manuscript and provided critical input regarding its intellectual content. All authors read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.