
Abstract
Background
B3GALNT2 mutations cause α-dystroglycanopathy (α-DGP), a rare condition characterized by muscular dystrophy, brain malformations, and developmental delay. However, its pathogenic mechanisms remain poorly understood. To date, limited cases have been reported, and the pathogenic mechanisms remain incompletely understood.
Methods
Clinical and genetic data from 3 newly diagnosed Chinese patients and 28 patients previously diagnosed with B3GALNT2-related α-DGP were analyzed. Using patient-derived fibroblasts, α-dystroglycan (α-DG) glycosylation and laminin-binding capacity were assessed by immunoblotting, laminin overlay and immunofluorescence. B3GALNT2 mRNA and protein levels were quantified by real-time PCR and immunoblotting. Enzymatic activity was measured using purified recombinant B3GALNT2 proteins. Differentially expressed genes were identified via an mRNA microarray.
Results
All three patients carried compound heterozygous variants involving one truncating and one missense mutation. Two novel mutations (c.657_658insTT and c.1384T > C) were identified. Functional studies confirmed that the missense mutations (Y436C and C462R) impaired enzymatic activity to 40–50% of wild-type levels, while splice variants caused frameshifts and likely complete loss of protein. Despite partial residual activity, all patients showed severely reduced α-DG glycosylation and loss of laminin binding, consistent with a functional threshold effect. Transcriptomic analysis revealed upregulation of CHST10 in two patients.
Conclusions
This study expands the mutational spectrum of B3GALNT2-related α-DGP and provides mechanistic insight into the pathogenicity of novel variants. Our findings support a functional threshold model for B3GALNT2 activity in α-DG glycosylation and suggest CHST10 as a potential transcriptional responder to glycosylation defects. These results deepen the understanding of B3GALNT2-related dystroglycanopathies and may inform future diagnostic and therapeutic strategies.
Background
α-Dystroglycanopathies (α-DGPs), also known as muscular dystrophy dystroglycanopathies (MDDGs), is a common subgroup of muscular dystrophies caused by hypoglycosylation of α-dystroglycan (α-DG). 1 The clinical spectrum of α-DGPs includes severe MDDG type A, intermediate MDDG type B, and mild MDDG type C. 2 MDDG type A presents as early-onset congenital muscular dystrophy (CMD) with brain and eye involvement, whereas MDDG type B manifests as CMD with or without mental retardation (MR). MDDG type C is characterized by late-onset limb-girdle muscular dystrophy (LGMD). MDDGs present with heterogeneous phenotypes and are caused by mutations in at least 19 genes, including DAG1, which is responsible for primary dystroglycanopathy, as well as 18 other genes encoding known or putative glycosyltransferases involved in α-DG glycosylation. 1 These mutations can result in complete or partial loss of enzyme function, and the clinical manifestation often depends on mutation type and zygosity—for example, recessive missense mutations may only cause disease in the homozygous state. Currently, no specific therapies are available to treat the underlying cause of α-DGPs. Further research is needed to understand the precise genetic and molecular basis of this disease. This knowledge can aid in the development of targeted interventions that address the underlying cause of α-DGPs.
α-DG is an extracellular peripheral membrane glycoprotein that binds to the transmembrane protein β-dystroglycan (β-DG) noncovalently. 3 The combination of these proteins with other transmembrane proteins (sarcoglycans α, β, γ, and δ and sarcospan) and intracellular proteins (dystrophin, syntrophin, dystrobrevin, and neuronal nitric oxide synthase) results in the formation of the dystrophin-glycoprotein complex (DGC). To date, 19 α-DGP causative genes have been discovered. Among them, DAG1 encoding dystroglycan, which is cleaved into α-DG and β-DG,1,4–6 is responsible for primary dystroglycanopathy when mutated. The remaining 18 genes encode glycosyltransferases involved in the post-translational modification of α-DG and are responsible for secondary dystroglycanopathies, in which the core protein is intact but hypoglycosylated due to defects in its enzymatic processing.7,8 A properly glycosylated α-DG binds laminin G-like domain-containing extracellular matrix proteins 9 such as laminin, 10 perlecan, 11 agrin, 12 and neurexin. 13 The functional glycan region of α-DG that binds extracellular matrix proteins is called matriglycan, which is a xylose (Xyl)-glucuronyl (GlcA) repeat disaccharide structure. The transmembrane protein β-DG associates with α-DG extracellularly and with dystrophin intracellularly. 14 Dystrophin binds to the cytoskeleton through interaction with F-actin. 15 The DGC acts as a bridge across the sarcolemma to connect the basal lamina of the extracellular matrix to the intracellular actin-based cytoskeleton 16 (Figure 1). This transmembrane link is essential for normal cellular functions, such as maintenance of structural integrity, signal transduction, and force transmission between the cytoskeleton and the extracellular matrix.17,18 In addition, the transmembrane link is required for neuromuscular junction formation; peripheral nerve myelination; neuronal migration in the brain; axon guidance; and development of the eye, brain, and other tissues.19–22 Mutations in the 19 causative genes impair the proper glycosylation of α-DG and disrupt the transmembrane link, ultimately leading to the occurrence of α-DGP diseases.
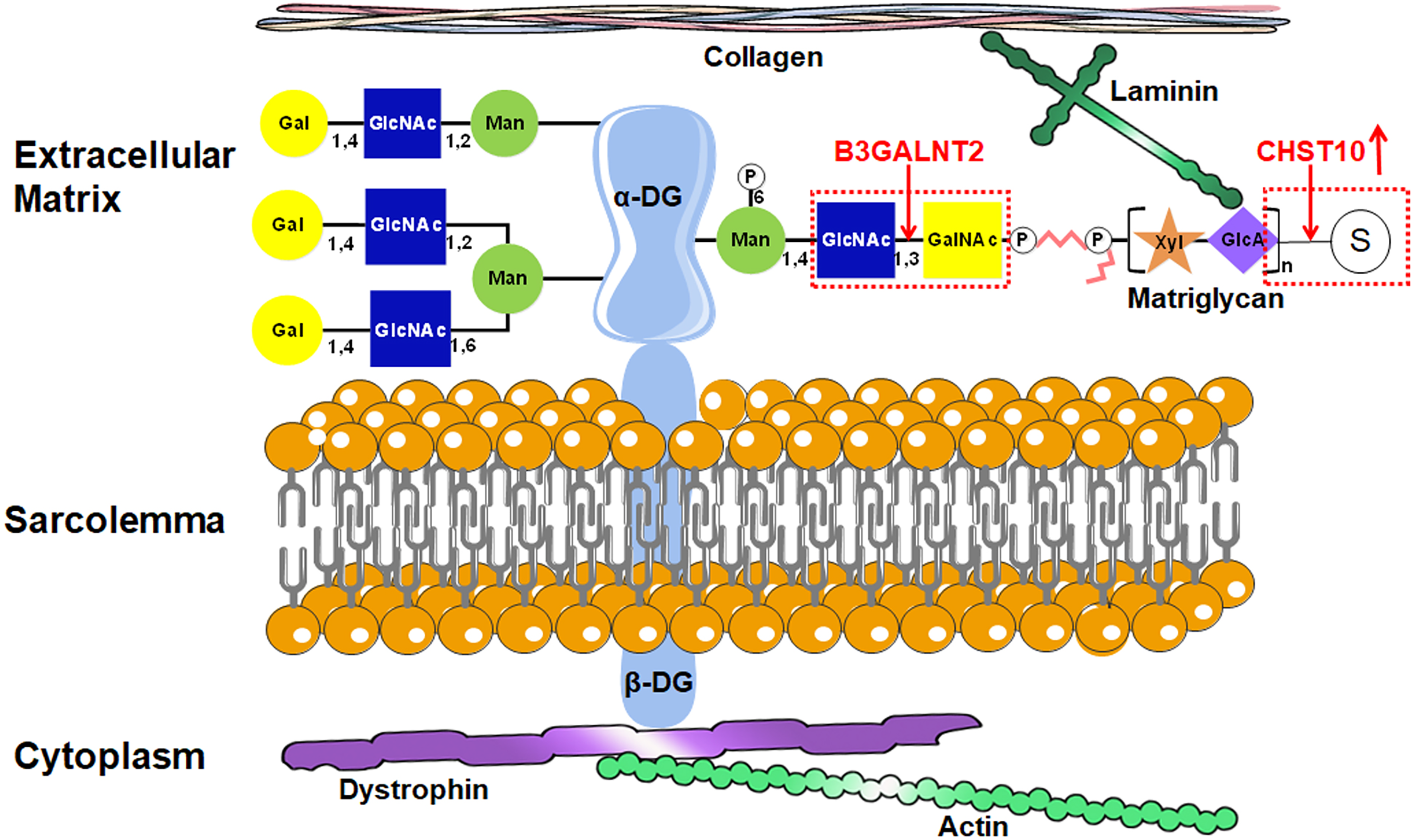
Schematic representation of the dystroglycan glycosylation pathway and the roles of B3GALNT2 and CHST10. The figure illustrates the structural organization of α-dystroglycan (α-DG) glycan moieties, their interaction with the extracellular matrix (ECM) components such as laminin, and the intracellular anchoring through β-dystroglycan (β-DG) to dystrophin and actin. B3GALNT2 is responsible for the β1,3 linkage of GalNAc to GlcNAc on the phosphorylated core M3 structure of α-DG in the endoplasmic reticulum (ER). CHST10, a Golgi-localized GalNAc 3-O-sulfotransferase, is depicted as upregulated in this study, although its direct functional contribution remains unclear.
The beta-1,3-N-acetylgalactosaminyltransferase 2 (B3GALNT2) gene, encoding a glycosyltransferase enzyme involved in α-DG glycosylation, is one of the 19 α-DGP causative genes, identified in 2013. 23 Only 28 patients with B3GALNT2-related α-DGP have been described in the literature to date, involving a total of 31 distinct pathogenic variants reported across 10 studies.23–32 B3GALNT2 is a type II transmembrane glycosyltransferase that localizes to the endoplasmic reticulum (ER). Its function is to add N-acetylgalactosamine (GalNAc) to the terminal N-acetylglucosamine (GlcNAc) residue of O-glycans, contributing to the elongation and diversification of O-glycan chains. This glycosylation is crucial for the proper structure and function of α-DG. However, the specific mechanisms by which mutations in the B3GALNT2 gene contribute to the pathogenicity of α-DGP are not yet fully understood.
In this study, we report three additional patients from unrelated Chinese families who harbor compound heterozygous mutations in B3GALNT2, including two novel variants (c.657_658insTT and c.1384T > C). Additionally, we followed up on two previously reported patients and conducted a comprehensive review of all 28 previously reported patients with B3GALNT2-related α-DGP, including their clinical, radiological, and genetic findings. Furthermore, we confirmed the pathogenicity of the B3GALNT2 mutations identified in our three patients by conducting functional analysis of the mutations and assessing their impact on α-DG glycosylation. This approach is crucial in establishing a clear understanding of how these specific B3GALNT2 mutations affect the glycosylation process of α-DG.
Methods
Patients
Three Chinese patients with B3GALNT2 mutations were recruited from Beijing Children's Hospital beginning in 2018. The patients (2 females and 1 male) from 3 unrelated nonconsanguineous families were all Han Chinese. The study protocol ([2022]-E-034-Y) was approved by the Medical Ethics Committee of Beijing Children's Hospital, Capital Medical University. Written informed consent was obtained from all patients’ parents, permitting enrollment in the study and the publication of medical data. Clinical data, including age of onset, clinical manifestations, laboratory tests, and brain magnetic resonance imaging (MRI) findings, were collected (Supplemental Table 1).
Whole-exome sequencing
The genomic DNA of 3 patients and their parents was extracted from whole blood using a DNA Extraction Kit (TIANGEN, Beijing, China), and the concentration was determined using a NanoDrop 2000 instrument (Thermo Fisher Scientific, Waltham, MA, USA). Libraries were prepared using the Illumina standard protocol. Whole-exome sequencing (WES) of the 3 probands and their parents was performed as previously described. 33
In silico analysis of B3GALNT2
B3GALNT2 sequences were obtained from UniProtKB (UniProtKB: Q8NCR0). The structures of the wild-type and mutant B3GALNT2 proteins were predicted using SWISS-MODEL (https://swissmodel.expasy.org/). The structural impact of the Y436C and C462R mutations was analyzed using PyMOL software. Residues 436 and 462 with nearby residues were illustrated. For a clear demonstration of the interresidue relationship, some residues were highlighted in different colors with the computed hydrogen bonds labeled.
Fibroblast culture
Fibroblasts from 3 patients with B3GALNT2-related α-DGP and 3 healthy controls were established from skin biopsies according to standard protocols. The skin fibroblasts were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, Invitrogen, Carlsbad, CA, USA) supplemented with 20% fetal bovine serum (FBS, Gibco, Invitrogen, Carlsbad, CA, USA) at 37 °C in a humidified cell incubator with 5% CO2.
Real-time PCR
Total RNA from fibroblast cell lines was isolated using the Direct-zol RNA Miniprep kit (TIANGEN, Beijing, China). RNA concentration and purity were measured spectrophotometrically with a NanoDrop ND-8000 instrument (Thermo Fisher Scientific, Waltham, MA, USA). Reverse transcription was performed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. Real-time PCR was conducted on a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with SYBR Green PCR Master Mix (TIANGEN, Beijing, China) to monitor fluorescence signals. Relative gene expression levels were normalized to the endogenous control GAPDH and analyzed via the 2ΔΔCt method. Primer sequences used for amplification are provided in Table 1.
Primers used in this study.
Allele-specific PCR and Sanger sequencing
The primers’ sequences in this study were designed using Primer Premier 5.0 (PREMIER Biosoft International, Palo Alto, CA, USA). The primer pair F1/R1 targets the wild-type cDNA sequence for conventional PCR amplification, while the mutation-specific primer pair F2/R2 (Table 1) was designed based on the mutant cDNA to achieve allele-specific amplification. All PCR reactions were performed on an Applied Biosystems Veriti 96-Well Thermal Cycler (Thermo Fisher Scientific, Waltham, MA, USA) for 30 cycles. Amplified products were separated by 2% agarose gel electrophoresis and visualized under UV transillumination. The PCR products were subsequently purified and subjected to bidirectional sequencing on an ABI 3730xl Automated DNA Sequencer (Thermo Fisher Scientific, Waltham, MA, USA). Raw sequencing data were analyzed for base calling and sequence alignment using Chromas software (Technelysium Pty Ltd, Brisbane, Australia).
Minigene construction and splicing analysis
An in vitro splicing assay was performed using a hybrid minigene system. A primer was designed to synthesize a genomic fragment of the B3GALNT2 gene containing exon 3 and parts of the neighboring introns. Genomic DNA fragments were amplified from Patient 1, who carried a heterozygous splicing mutation (c.261-2A > G), and cloned and inserted into the pEASY-T1 vector following the manufacturer's instructions. Both the wild-type and mutant alleles were detected and subsequently cloned and inserted into the pET01 Exontrap vector (Exontrap, MoBiTec GmbH, Germany). The wild-type and mutant minigene constructs were transiently transfected into HEK-293 T cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Following transfection, the cells were incubated for 48 h before total RNA extraction. The extracted RNA was then used to synthesize cDNA, which was subsequently amplified using specific primers designed for the pET01 Exontrap vector (forward: 5'-GATCGATCCGCTTCCTGCCCC-3’, reverse: 5'-GAGGTGGCCCGGCAG-3’).
WGA pull-down
Skin fibroblast lines from patients and healthy controls were harvested and lysed in lysis buffer (50 mM Tris-HCl, pH 7.6; 150 mM NaCl; 1% Triton X-100; 1 mM phenylmethylsulfonyl fluoride (PMSF)) for 30 min at 4 °C. The protein concentration was quantified using a Pierce™ BCA Protein Assay Kit (Thermo Scientific, USA). To enrich glycoproteins, equal amounts of protein were added to wheat germ agglutinin (WGA) agarose beads (Vector Labs) and incubated overnight at 4 °C. The beads were washed three times with ice-cold wash buffer (50 mM Tris-HCl, pH 7.6; 150 mM NaCl; 0.1% Triton X-100) and then boiled in 5× SDS-polyacrylamide gel electrophoresis (PAGE) loading buffer (3 min, 95 °C), followed by centrifugation at 14,000 rpm for 1 min. The eluted proteins were prepared for further analysis by SDS-PAGE and Western blotting.
Western blotting
To detect the expression and glycosylation levels of α-DG, WGA pull-down proteins were separated by 10% SDS-PAGE before being transferred to methanol-presoaked polyvinylidene fluoride (PVDF) membranes. The membrane was blocked with 3% bovine serum albumin (BSA) in KC buffer (100 mM NaCl, 20 mM Tris, pH 7.4) for 2 h at room temperature, followed by an overnight incubation at 4 °C with antibodies against the glycans of α-DG (IIH6C4 and VIA4-1) and the core protein of α-DG (ab151979). The signal was detected with an enhanced chemiluminescence (ECL) detection reagent after a 1 h incubation with a horseradish peroxidase (HRP)-conjugated secondary antibody. The immunoblot signals were quantified using ImageJ software.
To detect other proteins, including B3GALNT2, β-DG, CHST10, laminin and β-actin, fibroblasts from patients and healthy controls (HCs) were collected and lysed in RIPA buffer with a protease inhibitor cocktail (Roche Molecular Biochemicals, Mannheim, Germany) with three freeze (−80 °C) and thaw RT cycles. The lysate was centrifuged at 12,000 rpm for 5 min at 4 °C, and the supernatant was harvested as a total protein sample. The protein concentration was determined using a Pierce™ BCA Protein Assay Kit (Thermo Scientific, USA) following the manufacturer's instructions. The protein was separated and detected following the procedure described above using Tris-buffered saline with 0.1% Tween 20 detergent (TBST) buffer (50 mM Tris-HCl (pH 7.4), 0.9% NaCl, and 0.1% Tween 20). Information on the antibodies is shown in Table 2.
Primary antibodies used in this study.

Laminin overlay
WGA pull-down proteins were separated by 10% SDS-PAGE and then transferred to methanol-presoaked PVDF membranes. The membranes were blocked with 5% skim milk in laminin-binding buffer (LBB) (10 mM triethanolamine-HCl, 140 mM NaCl, 1 mM CaCl2, and 1 mM MgCl2, pH 7.6) at room temperature for 1 h and washed with LBB. The membrane was incubated with 7.5 nM mouse Engelbreth-Holm-Swarm (EHS) laminin (L2020, Sigma‒Aldrich, St Louis, MO, USA) at 4 °C for 16 h in LBB with 3% BSA. The membrane was washed and incubated with anti-laminin at room temperature for 3 h, and the signal was detected with an ECL detection reagent after a 1 h incubation with an HRP-conjugated secondary antibody.
Immunofluorescence staining
Fibroblasts at 60% confluence were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde (PFA) at room temperature for 10 min, washed with PBS, and then blocked in 10% goat serum in PBS without 0.1% Triton X-100 at room temperature for 1 h. The cells were incubated at 4 °C overnight with primary antibodies recognizing the glycans of α-dystroglycan (IIH6C4) and laminin, followed by incubation with secondary antibodies conjugated with either Alexa Fluor 488 (goat anti-mouse IgG H&L, Invitrogen cat. no.) A11001 or Alexa Fluor 647 (goat anti-rabbit IgG H&L, ab150079) at room temperature for 1 h in the dark. After primary and secondary antibody incubations, the samples were washed with PBS. Antifade mounting medium with DAPI (Beyotime Biotechnology, p0131) was used to stain the cell nuclei (blue).
mRNA microarray analysis
Fibroblasts from three patients and three age- and sex-matched HCs were collected. Total RNA was extracted and quantified with a NanoDrop instrument (Thermo Scientific). RNA integrity was assessed using an Agilent Bioanalyzer 2100 (Agilent Technologies). An Agilent SurePrint G3 Human Gene Expression v3 Microarray (8 × 60 K Design ID: 072363) was used for fibroblasts. Sample labeling, microarray hybridization and washing were performed according to the manufacturer's standard protocols. After washing, the arrays were scanned with an Agilent G2505C Scanner (Agilent Technologies). Feature Extraction software (version 10.7.1.1; Agilent Technologies) was used to analyze the array images to obtain raw data. Next, box plot analysis, Pearson correlation analysis, and principal component analysis (PCA) were performed to analyze the experimental data. Differentially expressed genes (DEGs) were then identified according to the criteria of fold change ≥2.0, P value ≤0.05, and functional relevance to the process of interest (glycosylation). The selected DEGs were then subjected to cluster analysis, Gene Ontology (GO) analysis, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis.
Expression and purification of B3GALNT2
The construct expressing B3GALNT2 without its transmembrane region was synthesized corresponding to human B3GALNT2 (NM_152490) cDNA with a sequence encoding an N-terminal 10×His tag. This wild-type B3GALNT2 plasmid was used as a template for site-directed mutagenesis PCR to introduce the identified mutations c.1307A > G and c.1384T > C, generating the corresponding mutant B3GALNT2 cDNA sequences. The synthesized wild-type plasmid and two plasmids with point mutations were subsequently separately transfected into HEK293 cells. Transfection was performed using Sinofection transfection reagent (STF02, Sino Biological) according to the manufacturer's instructions. The transfected cells were cultured in serum-free medium (SMM, 293-TI, Sino Biological) and maintained in Erlenmeyer flasks on an orbital shaker at 37 °C with sufficient stirring for 6 d to facilitate protein expression. The B3GALNT2 protein (amino acids 24-500) secreted by the transfected HEK293E cells was purified using Ni Sepharose Excel (17371206, Cytiva) following the manufacturer's instructions. The purity of the protein was confirmed by SDS-PAGE and Coomassie brilliant blue staining.
Enzyme activity analysis
The enzyme activity of B3GALNT2 was assessed using a glycosyltransferase activity kit (EA001, R&D Systems) and specific substrates. The kit included uridine diphosphate (UDP)-GalNAc (U5252, Sigma) as the donor substrate and benzyl-GlcNAc (SC-221296, Santa Cruz Biotechnology) as the acceptor substrate. The kit incorporated a coupling phosphatase that was used to remove inorganic phosphate from the remaining UDP molecules generated during glycosyltransferase reactions. To assess B3GALNT2 activity, the released inorganic phosphate was detected using malachite green phosphate-detecting reagents. The rate of inorganic phosphorus production, which reflects the enzymatic activity of B3GALNT2 in a given protein mixture, was then measured. The reaction conditions and detailed methods used in the assay were according to the manufacturer's protocol provided with the glycosyltransferase activity kit.
Statistical analysis
Unpaired Student's t test was used for statistical analysis. The data are presented as the means ± standard errors of the means. Statistical analysis was performed using GraphPad Prism software (GraphPad Software, San Diego, CA, USA), and statistical significance was denoted as *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Results
B3GALNT2 mutations in three unreported patients
We identified compound heterozygous mutations in B3GALNT2 in three unrelated patients, each carrying one allele predicted to disrupt protein synthesis (splicing or frameshift) and one missense variant located within the C-terminal galactosyltransferase domain. Specifically, Patient 1 (P1) harbored c.1307A > G (p.Y436C) and c.261-2A > G, Patient 2 (P2) had a novel c.1384T > C (p.C462R) variant and c.48dupG, and Patient 3 (P3) carried the recurrent c.979G > A (p.D327N) along with a novel c.657_658insTT variant (Figure 2a). All variants and their inheritance were confirmed by Sanger sequencing (Supplementary Figure 1a-c).
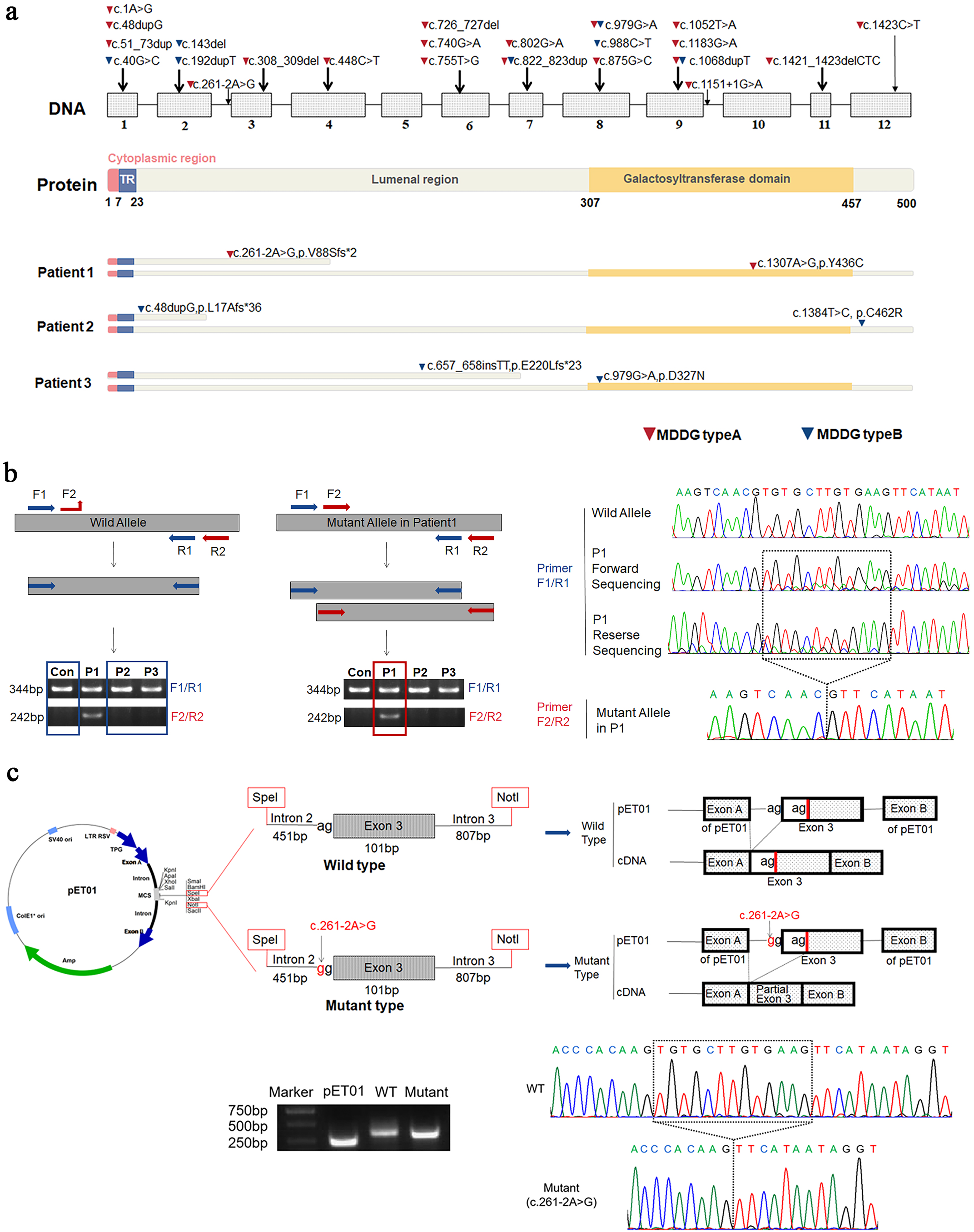
Identification of compound heterozygous B3GALNT2 mutations in three unrelated patients. (a) Schematic overview of all known B3GALNT2 mutations with their exon locations and protein domain structure. Mutations identified in this study are indicated below for each patient, with triangles marking MDDG type A (red) and type B (blue) variants. (b) RT-PCR and allele-specific PCR analysis demonstrating a 13-bp deletion in exon 3 caused by the c.261-2A > G splice-site mutation in Patient 1. (c) Minigene splicing assay confirming the exon 3 skipping event in the mutant allele.
To explore the splicing consequence of c.261-2A > G, RT-PCR and allele-specific PCR performed on patient-derived fibroblasts revealed a 13 bp deletion in exon 3 (Figure 2b), which was further validated using a minigene splicing assay (Figure 2c). These results support that the splicing defect causes a frameshift and likely triggers nonsense-mediated mRNA decay.
Impact of mutations on B3GALNT2 function
Quantitative PCR and Western blot analyses revealed no significant differences in B3GALNT2 mRNA or protein expression between patients and controls (Figure 3a-c), suggesting that the observed pathogenicity is primarily due to loss of enzymatic function rather than reduced expression.
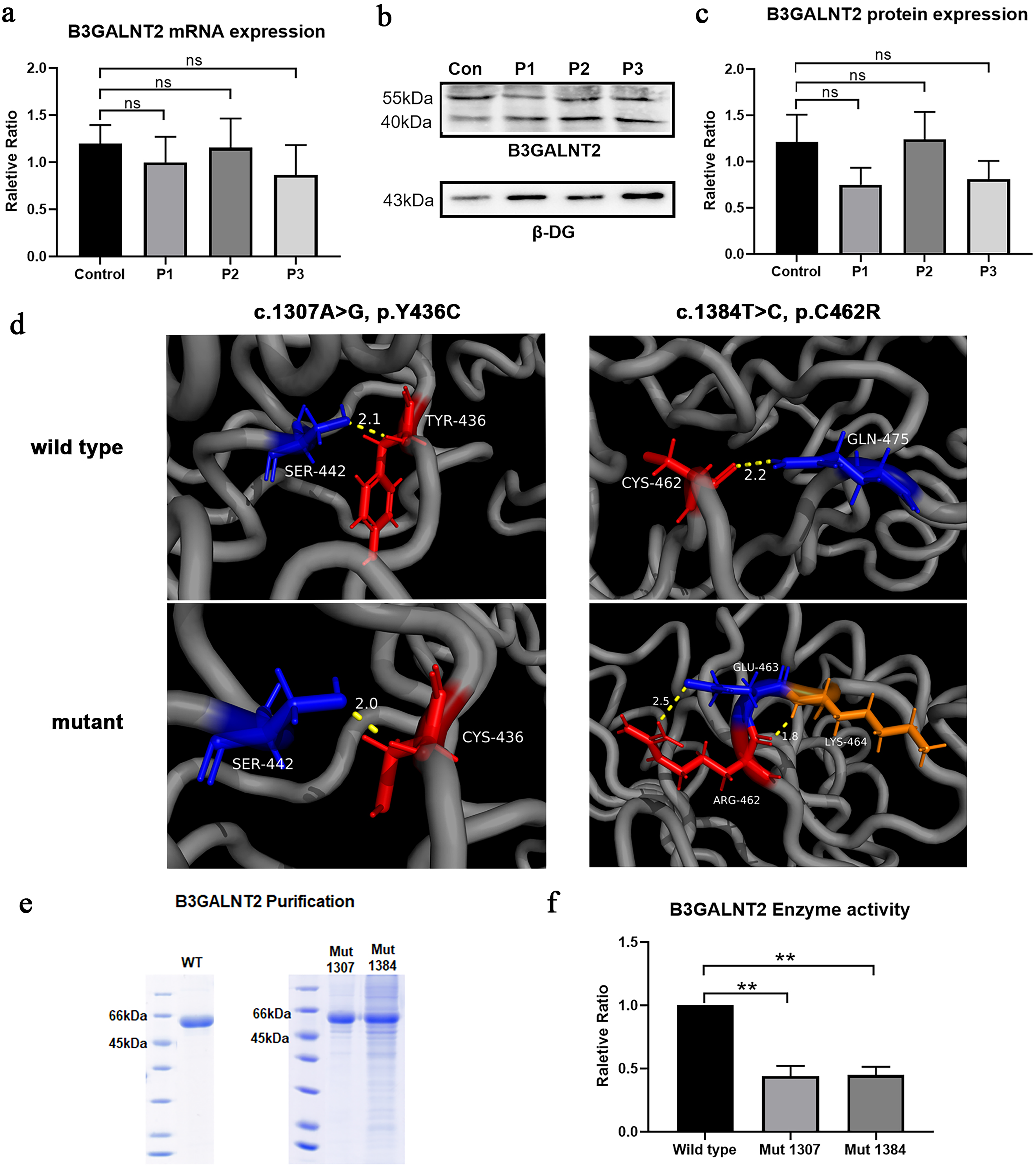
Effects of B3GALNT2 variants on gene expression, protein structure, and enzymatic activity. (a–c) Quantitative RT-PCR and Western blot analyses revealed no statistically significant differences in B3GALNT2 mRNA or protein expression levels between controls and patients. However, a downward trend was observed in Patient 1 (P1) and Patient 3 (P3), with mRNA levels slightly decreased and protein levels reduced by approximately 40%, suggesting that partial post-transcriptional instability may contribute alongside functional impairment. (d-left) In the wild-type structure, the distance between residue Tyr436 (tyrosine, red) and the neighboring Ser442 (serine, blue) is 2.1 Å, indicated by a yellow dashed line. Tyr436 is mutated to Cys436 (cysteine, red), introducing a new thiol-containing residue. The interatomic distance between Cys436 and Ser442 slightly decreases to 2.0 Å; however, this substitution may facilitate the formation of aberrant disulfide bonds (if other cysteine residues are nearby), potentially disrupting normal protein folding. Furthermore, the polarity shift introduced by the mutation may weaken the original hydrogen bond interaction, rendering this region more susceptible to conformational perturbation. (d-right) In the wild-type model, CYS-462 (red) and GLN-475 (blue) are positioned in close proximity with a distance of 2.2 Å, which may support a stable disulfide bond or non-covalent interaction, as shown by the yellow dashed line. Upon mutation to ARG-462 (arginine, red), a bulky and positively charged side chain is introduced. The mutant ARG-462 interacts differently with surrounding residues: it lies 2.5 Å from GLU-463 (blue), possibly generating electrostatic stress or interaction, and only 1.8 Å from LYS-464 (orange), another positively charged residue. This close distance between like-charged residues may induce charge repulsion. The resulting structural disruption likely destabilizes the local conformation and impairs the catalytic domain of B3GALNT2. (e) SDS-PAGE analysis of purified recombinant B3GALNT2 proteins (wild-type, Y436C, and C462R mutants), confirming comparable expression and successful purification. (f) Enzymatic activity assays showed that both Y436C and C462R mutants retained approximately 40–50% of wild-type catalytic activity.
We selected the two missense variants identified in P1 and P2 (Y436C and C462R) for further analysis, as functional data for these variants were previously lacking and C462R represents a novel mutation. In contrast, the p.D327N variant found in P3 has been frequently reported as a recurrent pathogenic mutation in B3GALNT2; therefore, additional functional validation was not performed.
Structural modeling indicated that the Y436C mutation may introduce a cysteine capable of forming non-native disulfide bonds, potentially disturbing proper protein folding. Conversely, the C462R mutation is predicted to disrupt a native disulfide bond, destabilizing the local structure (Figure 3d). Enzymatic activity assays using purified recombinant proteins showed that both Y436C and C462R mutations retained approximately 40–50% of the wild-type enzymatic activity (Figure 3e-f), indicating partial catalytic function.
To determine whether reduced B3GALNT2 activity affects α-dystroglycan (α-DG) glycosylation, we examined patient fibroblasts. Western blot analysis revealed a marked reduction in glycosylated α-DG levels using the IIH6C4 and VIA4-1 antibodies (Figure 4a-b), while the expression of the α-DG core protein and β-DG remained unchanged (Figure 4a, 4c). Immunofluorescence in three patients’ fibroblasts demonstrated a loss of IIH6 reactivity and disrupted colocalization with laminin (Figure 4d and Supplementary Figure 2). Consistently, laminin overlay assays confirmed a complete loss of α-DG laminin-binding ability in all three patients (Figure 4e).
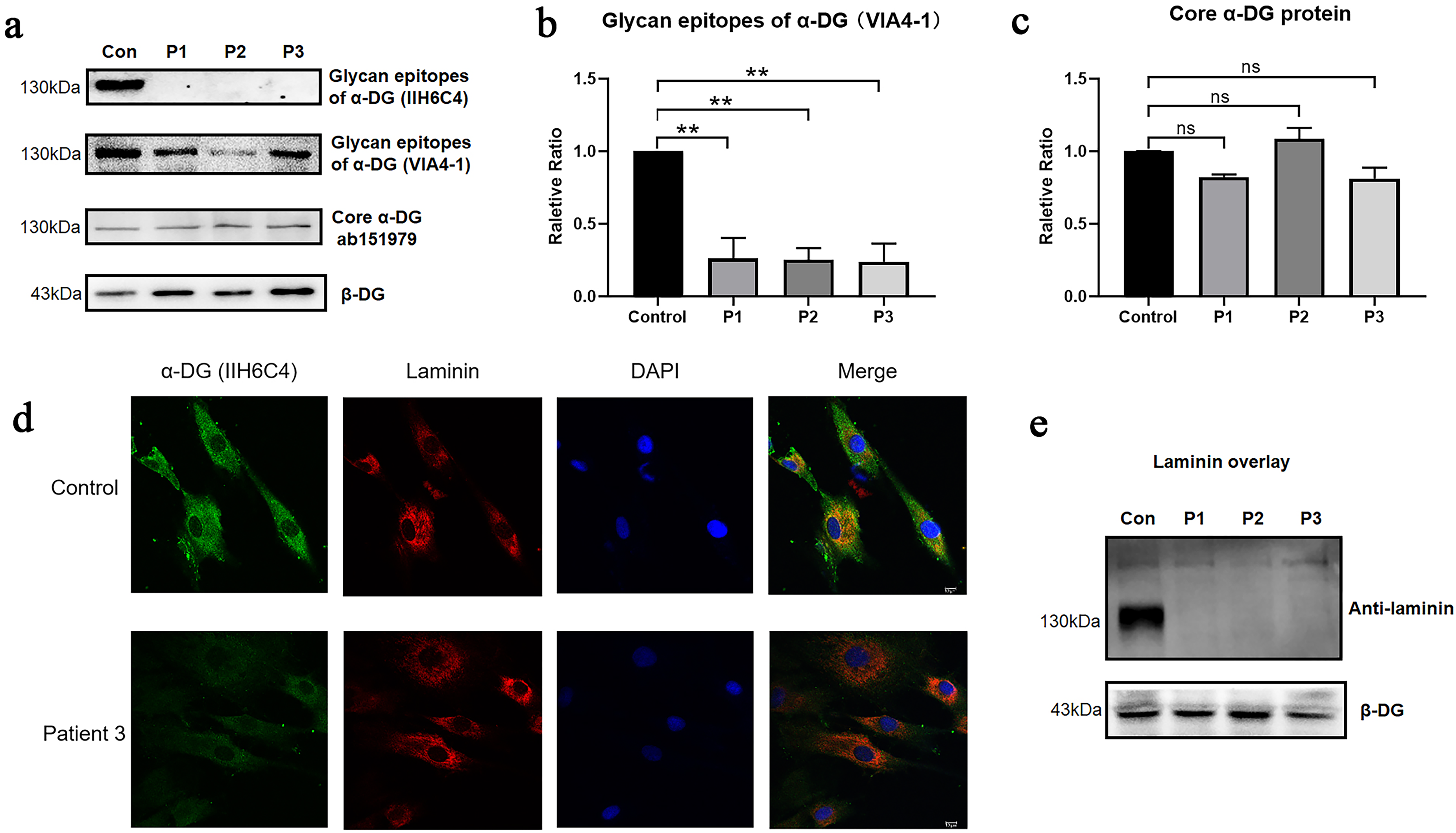
α-DG glycosylation level and laminin-binding activity. (a) Western blot analysis shows markedly reduced IIH6C4 and VIA4-1 reactivity in patient-derived fibroblasts, indicating hypoglycosylation of α-DG, while core α-DG and β-DG expression remain unchanged. (b–c) Quantification of glycan epitope and core α-DG levels. (d) Immunofluorescence staining in Patient 3 fibroblasts demonstrates loss of IIH6 signal and absence of α-DG–laminin colocalization compared with controls. (e) Laminin overlay assay confirms complete loss of α-DG laminin-binding capacity in all three patients.
Although the mutant enzymes retained 40–50% activity, laminin binding was completely abolished in all patients. This raised the question of whether similar biochemical defects translate into comparable clinical phenotypes. To address this, we further analyzed the genotype–phenotype correlation.
Genotype-phenotype correlation analysis
Clinically, P1 presented with MDDG type A (muscle-eye-brain disease) characterized by brainstem malformation and cerebellar atrophy, while P2 and P3 exhibited MDDG type B with relatively milder white matter abnormalities and cerebellar T2 hyperintensity. P3 also showed polymicrogyria and ventricular enlargement (Figure 5a). Detailed clinical features of the three patients are provided in Supplementary Table 1.
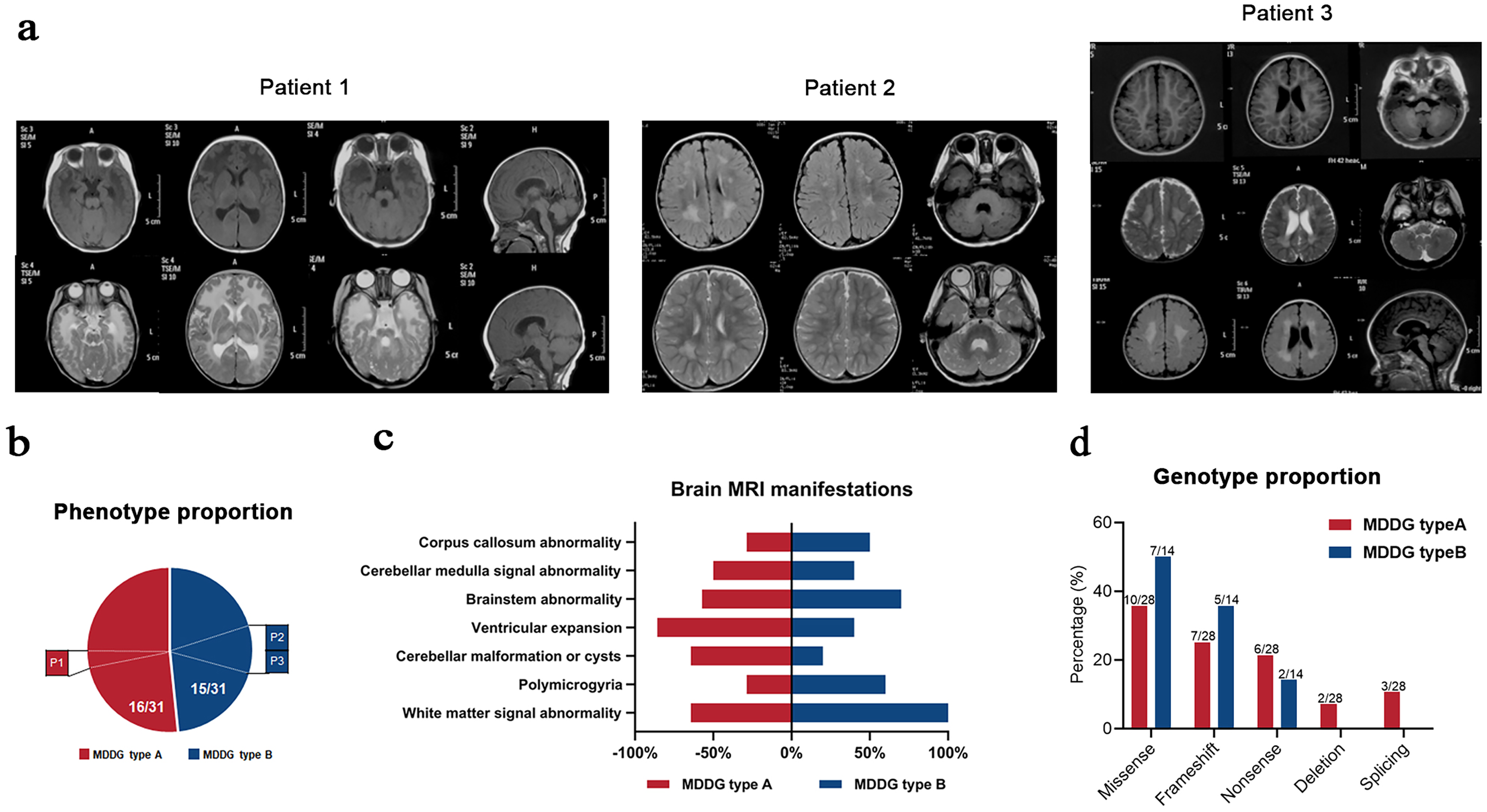
Phenotype–genotype correlation in B3GALNT2-related α-DGP. (a) Representative brain MRI findings in three patients. P1 shows brainstem malformation and cerebellar atrophy (MDDG type A), whereas P2 and P3 present milder features including white matter signal abnormalities, cerebellar T2 hyperintensity, and in P3, polymicrogyria and ventriculomegaly (MDDG type B). (b) Phenotypic classification of 31 total cases (28 literature-reported and 3 current) into MDDG types A and B. (c) Brain MRI abnormality frequency distribution in MDDG types A vs. B. (d) Genotype distribution by mutation type between the two clinical subgroups.
Supplemental Table 1. Genotype and Phenotype of 31 B3GALNT2-related DGP patients (Parts 1 and 2)
Among all 31 reported B3GALNT2-related α-DGP cases (28 from the literature and 3 from this study), 16 were classified as MDDG type A and 15 as type B (Figure 5b). All patients presented with motor and cognitive developmental delay, and speech delay or impairment was a common clinical feature in all patients except for individuals whose language abilities were not assessed (Supplemental Table 1). In addition, all patients who underwent brain MRI presented distinct brain abnormalities, with white matter abnormalities being the most prominent feature (Figure 5c).
A total of 26 distinct B3GALNT2 mutations were identified across the 31 cases, including 11 missense variants, 8 frameshift variants, 5 nonsense variants, 1 in-frame deletion, and 1 splice-site mutation (Figure 2a). Of particular interest, the missense mutation c.979G > A (p.D327N) was observed in nine patients—one Swedish, five from an Iranian family, and three from unrelated Chinese families—accounting for 29.0% (9/31) of all reported cases. This highlights c.979G > A as the most frequently recurrent mutation in B3GALNT2-related α-DGP.
When stratified by clinical severity, nonsense and splice-site mutations were more commonly observed in patients with MDDG type A, while missense and frameshift mutations predominated in those with MDDG type B (Figure 5d). Nevertheless, no strict genotype–phenotype correlation could be established, suggesting the involvement of additional modifiers.
Transcriptomic profiling and CHST10 expression analysis
To explore potential downstream effects and modifiers associated with B3GALNT2 deficiency, we performed transcriptome profiling on fibroblasts derived from the three patients. Differential gene expression analysis identified 192 upregulated and 159 downregulated transcripts compared to controls. Among the glycosylation-related genes, 12 were upregulated and 11 downregulated in patient samples (Figure 6a). KEGG pathway enrichment analysis revealed significant enrichment in the “mannose-type O-glycan biosynthesis” pathway (Figure 6b), with CHST10 being the only gene consistently upregulated within this category.
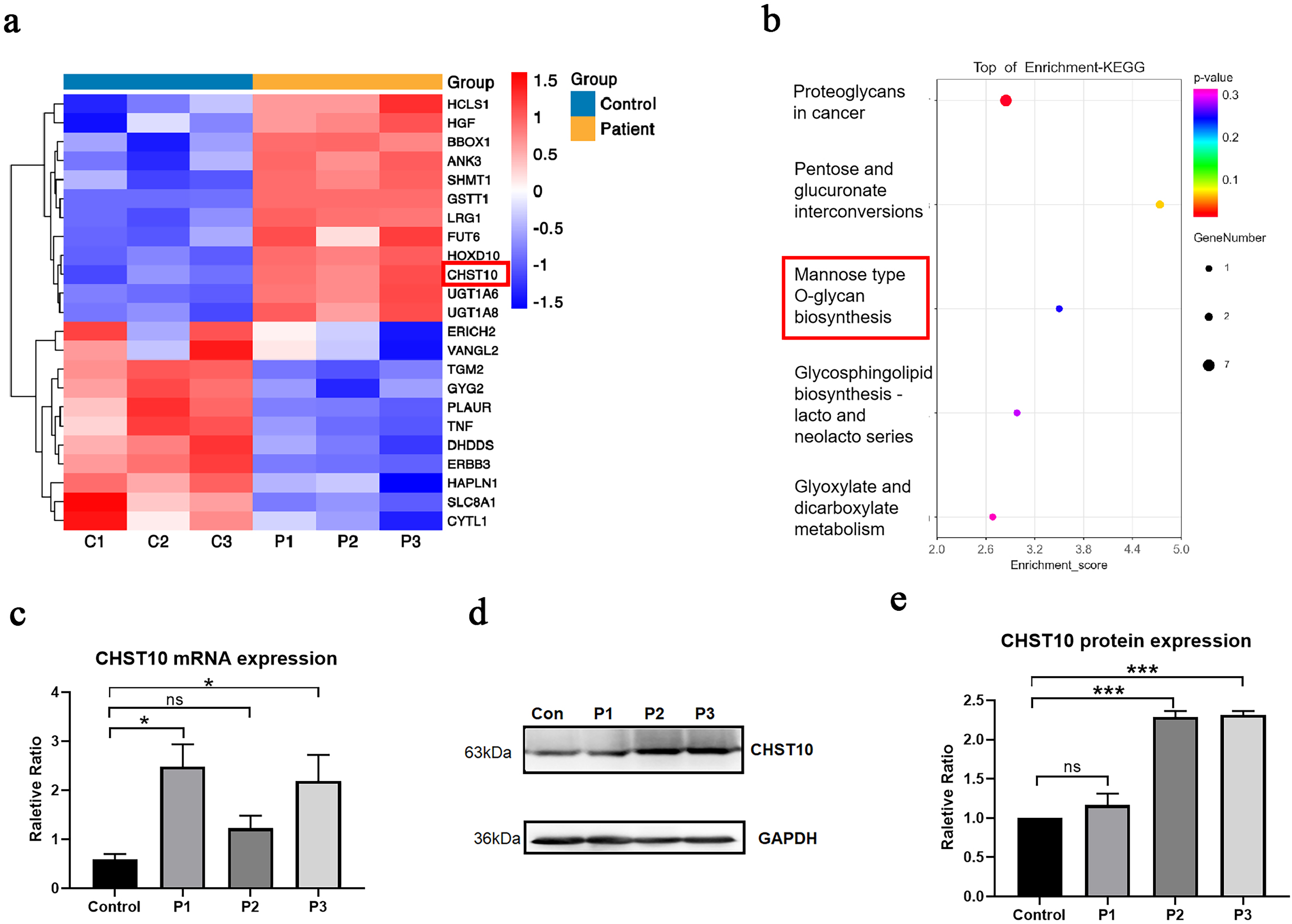
Transcriptomic profiling reveals CHST10 upregulation in patient fibroblasts. (a) Heatmap of differentially expressed glycosylation-related genes in fibroblasts from three B3GALNT2-mutant patients (P1–P3) compared with three healthy controls (C1–C3). A total of 23 genes involved in glycosylation were differentially expressed, including 12 upregulated and 11 downregulated genes. CHST10 was one of the genes consistently upregulated across all three patients. (b) KEGG enrichment analysis of glycosylation-related DEGs revealed significant enrichment in the “mannose-type O-glycan biosynthesis” pathway. CHST10 was the only gene upregulated in this pathway. (c) Quantitative real-time PCR confirmed elevated CHST10 mRNA expression in P1 and P3, although P2 did not show statistically significant upregulation. (d-e) Western blot analysis revealed inter-individual variation in CHST10 protein expression. Densitometric quantification of CHST10 protein levels normalized to GAPDH confirmed significant upregulation in P2 and P3 compared to control, while P1 remained unchanged.
CHST10 mRNA expression was evaluated by quantitative real-time PCR. Although upregulation was observed in P1 and P3, no significant increase was detected in P2 (Figure 6c). Immunoblot analysis further revealed inter-individual variation at the protein level, with no obvious increase in CHST10 protein expression in P1, despite elevated mRNA levels (Figure 6d-e).
These findings indicate that CHST10 is transcriptionally upregulated in a subset of patients with B3GALNT2 mutations, potentially reflecting a secondary glycosylation-related response. Further investigation is needed to clarify its biological significance and mechanistic contribution to α-dystroglycanopathy.
Discussion
In this study, we systematically reported three Chinese patients harboring compound heterozygous mutations in B3GALNT2 and investigated the underlying pathogenic mechanisms and potential molecular regulatory factors through an integrated approach involving molecular genetics, protein structural modeling, biochemical functional assays, and transcriptomic profiling. We identified six pathogenic variants, including two previously unreported mutations (c.657_658insTT and c.1384T > C), thereby expanding the mutational spectrum of B3GALNT2-related α-dystroglycanopathy (α-DGP).
B3GALNT2 encodes a monomeric β1,3-N-acetylgalactosaminyltransferase localized in the endoplasmic reticulum. It has only one well-characterized transcript, NM_152490.5, with confirmed functional evidence, while the alternative transcript NM_001277155.3 has not yet been functionally characterized. 34 B3GALNT2 catalyzes the synthesis of the GalNAc-β1,3-GlcNAc structure and serves as a rate-limiting enzyme in the biosynthesis of functional O-glycans on α-dystroglycan (α-DG).1,34
All three patients in our study carried compound heterozygous variants, typically with one allele harboring a splice site or frameshift mutation that is likely to result in truncated or degraded protein, and the other allele carrying a missense mutation within or near the catalytic domain. Expression analyses showed no statistically significant difference in B3GALNT2 mRNA and protein levels between patients and controls overall; however, patients P1 and P3 exhibited a ∼40% reduction in protein levels, suggesting that even a moderate decrease in enzyme abundance may impact glycan synthesis due to the rate-limiting nature of B3GALNT2.2,25
The c.979G > A (p.Arg327Gln) mutation carried by P3 is a recurrent hotspot variant that has been previously associated with typical α-DGP phenotypes.24,27 In contrast, P1 and P2 carried two missense variants with limited prior characterization, including the novel variant c.1384T > C. Structural modeling revealed that the Y436C mutation may introduce an aberrant cysteine residue capable of forming non-native disulfide bonds and disrupting local folding, while the C462R substitution may disrupt the native Cys462–Glu463 disulfide bridge and introduce a positively charged residue that likely repels adjacent Lys464, thus destabilizing the catalytic domain. 34 These structural disruptions are consistent with our biochemical findings showing a 50–60% reduction in enzymatic activity compared to wild-type.
In contrast, the pathogenic mechanism of the splice-site mutation c.261-2A > G is likely mediated through aberrant mRNA splicing and frameshift-induced premature termination. cDNA sequencing confirmed a 13 bp deletion leading to a premature stop codon, consistent with nonsense-mediated mRNA decay (NMD) and complete loss of B3GALNT2 function. 34 Functional assays confirmed that such truncating mutations are associated with a severe reduction in α-DG glycosylation and complete loss of laminin-binding capacity, underscoring the pathogenic significance of total protein loss.
Despite partial residual enzymatic activity in some missense variants, all patients exhibited markedly reduced α-DG glycosylation and a complete loss of laminin-binding activity. These findings support the existence of an “activity threshold” model, whereby B3GALNT2 enzymatic function must exceed a critical threshold to support the elongation of functional glycan structures on α-DG.1,7,18 This observation further highlights that enzyme activity assays reflect intermediate biochemical steps, whereas laminin-binding assays measure the final cell-level functional outcome, thus providing complementary but distinct layers of functional insight.
Phenotypic variability among patients also merits attention. P1 exhibited the more severe MDDG type A phenotype, while P2 and P3 were consistent with milder MDDG type B presentations. Previous studies suggest that truncating mutations are more often associated with severe phenotypes, while missense or frameshift mutations are frequently linked to milder presentations.25–27 However, this correlation is not absolute, implying that phenotypic severity may be influenced by additional factors such as genetic background, tissue-specific expression, or compensatory modulation within the glycosylation network.25,28
To explore possible transcriptional regulators, transcriptomic analysis revealed significant upregulation of CHST10 in patients. CHST10 encodes a Golgi-localized GalNAc-3-O-sulfotransferase that catalyzes 3-O-sulfation of glucuronic acid on matriglycan at the terminal end of α-DG O-glycans. Previous studies suggest that this sulfation may inhibit further glycan elongation and serve as a negative regulator of α-DG glycosylation.35–37 However, in our study, although mRNA levels of CHST10 were elevated (notably in P1), the protein level remained unchanged, suggesting a possible non-specific or compensatory transcriptional response rather than a primary pathogenic mechanism.
Moreover, as CHST10 is predominantly expressed in neural tissue 36 and no studies to date have reported its dysregulation in secondary dystroglycanopathies, we propose that its upregulation reflects an indirect or compensatory response to B3GALNT2 dysfunction. Whether CHST10 contributes to the fine-tuning of α-DG glycan structure remains to be clarified. Due to the slow proliferation of patient-derived fibroblasts following shRNA-mediated CHST10 knockdown, we were unable to perform functional validation of its role.
In conclusion, our study provides comprehensive evidence that B3GALNT2 mutations impair α-DG glycan synthesis either by reducing enzyme activity or abolishing protein expression. We propose a “functional threshold” model for enzymatic activity required to maintain α-DG functionality and identify CHST10 as a potential secondary transcriptional modulator in the glycosylation network. These findings broaden the mutational and mechanistic understanding of B3GALNT2-related α-DGP and offer insights for future research and therapeutic development.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602251360270 - Supplemental material for Pathogenic mechanisms and clinical insights into B3GALNT2-related alpha-dystroglycanopathies
Supplemental material, sj-docx-1-jnd-10.1177_22143602251360270 for Pathogenic mechanisms and clinical insights into B3GALNT2-related alpha-dystroglycanopathies by Xiaona Fu, Hui Wang, Wenjia Chai, Xiaoyu Chen, Danyu Song, Wei Wang, Jingwei Zhong, Zhimei Liu, Xiao Tong, Hui Xiong, Xiaotun Ren and Jingang Gui in Journal of Neuromuscular Diseases
Supplemental Material
sj-docx-2-jnd-10.1177_22143602251360270 - Supplemental material for Pathogenic mechanisms and clinical insights into B3GALNT2-related alpha-dystroglycanopathies
Supplemental material, sj-docx-2-jnd-10.1177_22143602251360270 for Pathogenic mechanisms and clinical insights into B3GALNT2-related alpha-dystroglycanopathies by Xiaona Fu, Hui Wang, Wenjia Chai, Xiaoyu Chen, Danyu Song, Wei Wang, Jingwei Zhong, Zhimei Liu, Xiao Tong, Hui Xiong, Xiaotun Ren and Jingang Gui in Journal of Neuromuscular Diseases
Footnotes
Abbreviations
Acknowledgments
We are grateful to the doctors and the patients and their families for their participation in this study. We greatly thank Prof. Francesco Muntoni and Dr. Silvia Torelli for their guidance with the immunoblotting experiments used to measure the degree of glycosylation of α-DG. We greatly thank Prof. Tatsushi Toda, Prof. Kazuhiro Kobayashi, and Prof. Motoi Kanagawa for their guidance with the laminin overlay experiment.
Ethical considerations
The study protocol ([2022]-E-034-Y) was approved by the Medical Ethics Committee of Beijing Children's Hospital, Capital Medical University, National Center for Children's Health (Beijing, China).
Consent to participate
Written informed consent for routine and investigative studies was obtained from the parents of all the patients.
Author contributions
The project was conceived and supervised by J.G., X.R. and H.X. With the help of H.W. and W.C., X.F. performed the in silico analysis of B3GALNT2, WGA pull-down, Western blotting, laminin overlay, expression and purification of B3GALNT2, enzyme activity analysis, and mRNA microarray analysis. H.W. performed the allele-specific PCR, Sanger sequencing, minigene construction, splicing analysis, and immunofluorescence staining. W.C. performed the fibroblast culture, real-time PCR and data analysis. X.C. and D.S. performed patient enrollment. H.X. provided help in the clinical diagnosis of the patients. W.W., J.Z., X.T. and Z.L. performed specimen sampling. X.F., H.W. and W.C. wrote the manuscript. All the authors read and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Key R&D Program of China (No. 2022YFC2703100), Joint Basic-Clinical Laboratory of Pediatric Epilepsy and Cognitive Developmental (No. 3-1-013-03), National Natural Science Foundation of China (No. 82173084, No. 82171393 and No. 82471430), Beijing Natural Science Foundation (No. 7224327 and No. 7234364), and Funding for Reform and Development of Beijing Municipal Health Commission.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Authors’ information
Per journal guidelines, this optional section can be included to list any relevant information about the authors that may aid the reader's interpretation of the article and understanding of the authors’ viewpoint.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.