
Abstract
The TNNC2 gene is crucial for skeletal muscle function, and pathogenic variants have been linked to congenital myopathies characterized by hypotonia, muscle weakness, and respiratory insufficiency. To date, TNNC2-related myopathies have been associated only with autosomal dominant missense variants. We report here the first family case of a recessive form of myopathy related to TNNC2. We identified the homozygous splice variant TNNC2(NM_003279.3):c.314 + 1G > C p.(?) in two siblings with a severe clinical presentation, resulting in one neonatal death and one medical termination of pregnancy. This variant induces a splicing defect that leads to a complete loss of the TNNC2 physiological transcript. This case expands the spectrum of TNNC2 variants with a late-onset fetal loss. To the best of our knowledge, this is the first case reporting a recessive form of severe neonatal hypotonia due to TNNC2 variant.
Background
The TNNC2 gene encodes the troponin C type 2 protein, expressed in fast twitch skeletal muscle fibers. This subunit of the troponin complex plays a major role in the striated muscle function as it binds calcium and subsequently reduces the steric hindrance of myosin heads allowing sarcomere contraction. 1 Pathogenic variants in TNNC2 have been associated with a form of congenital myopathy characterized by muscle weakness, hypotonia and respiratory insufficiency, along with various contractures, scoliosis, ptosis and retrognathia (MIM #620161). To this day, inheritance of this myopathy has exclusively been described with an autosomal dominant transmission of missense variants in TNNC2.
We report here the case of two siblings bearing a homozygous variant in the TNNC2 gene, from a family native from South Italia with no known history of consanguinity. To the best of our knowledge, this is the first case reporting a homozygous variant in TNNC2 associated with this phenotype, and suggesting a possible recessive form of TNNC2-related myopathy.
Case report
The family proband was a boy born preterm at 32 weeks of gestational age (GA), facing global hypotonia and no spontaneous breathing. Weight and height were respectively of 1640 g (37th percentile) and 43 cm (59th percentile). Cranial perimeter was of 32 cm (89th percentile). Pregnancy was marked by a severe hydramnios that first challenged physicians for esophageal atresia. At birth, an intubation was needed for resuscitation and physicians were unable to wean the neonate from the ventilator. Neurological exams revealed the absence of swallowing reflex along with a global hypotonia. Normal cerebral MRI excluded a central cause of hypotonia. CK levels reached at 990 U/L [39–308] during the first week of life. X-Ray showed thoracolumbar scoliosis with hypercyphosis and thin ribs (Figure 1A). The evaluation also revealed bilateral cryptorchidism. Pluridisciplinary and parental decision was to withdraw the care after 45 days of hospitalization without clinical improvement.
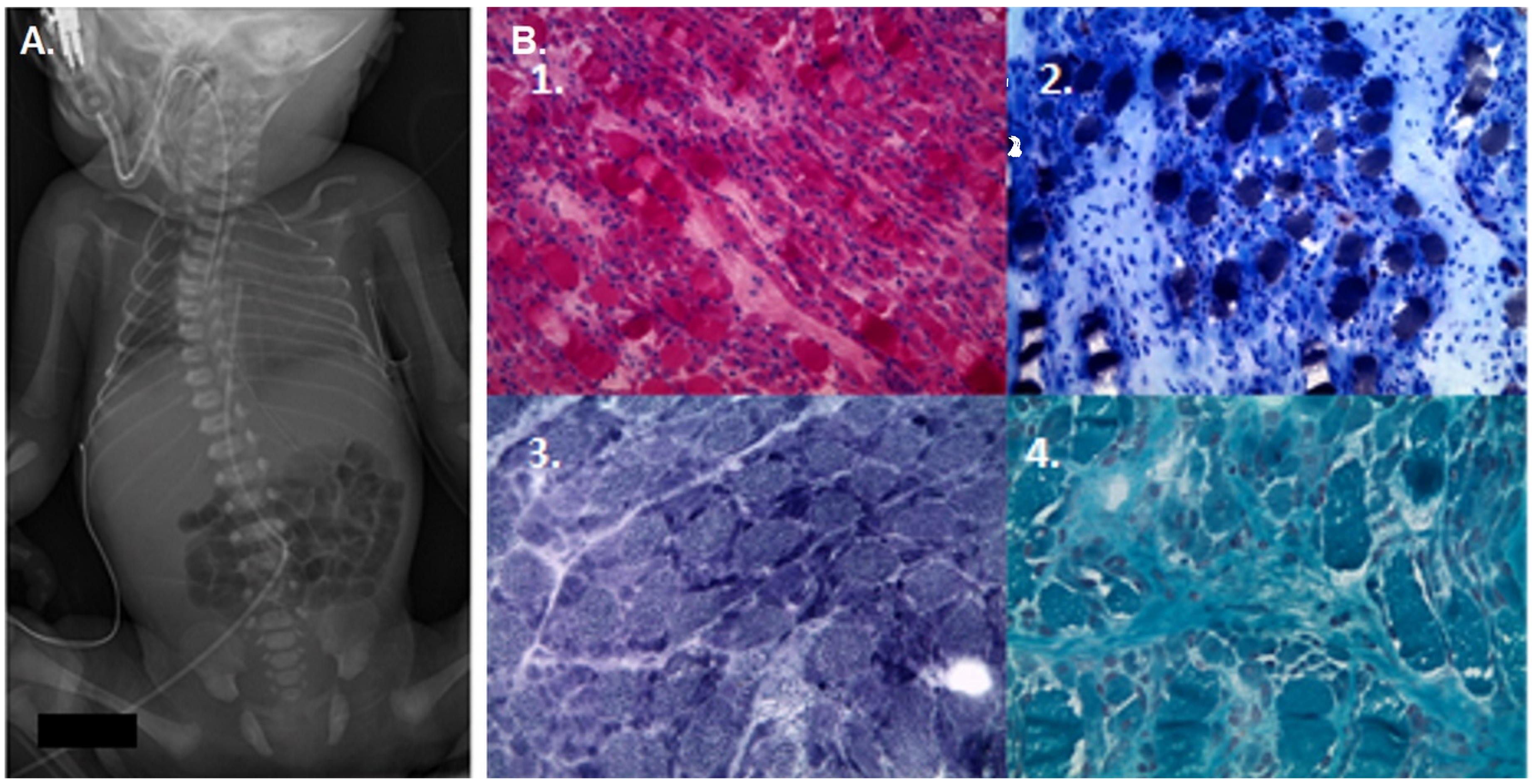
Imaging and histological finding at birth in the first affected child. A. Frontal X-Ray radiography at birth evidencing a thoracolumbar scoliosis with hypercyphosis along with thin pairs of ribs. B. Histological sections and stains from muscle biopsy on quadriceps vastus medialis. 1. Hematoxylin and eosin (H&E) and 2. ATPase pH 4.35 stainings show hypertrophic type I fibers (dark) and atrophic type II fibers (light). 3. NADH and oxidative technique and 4. Trichrome showed no abnormality.
A muscle biopsy was performed and revealed atrophied type II fibers along with type I fibers hypertrophy on hematoxylin and eosin (HE) and ATPase staining. Histoenzymatic techniques did not reveal any congenital myopathies hallmarks, such as cores or nemaline bodies. Immunohistochemical examination did not reveal any deficiency in proteins involved in muscular dystrophies (Figure 1B). No genetic cause was identified after next generation sequencing on patient's DNA using a panel of 128 genes involved in congenital myopathies and hypotonia. 2 Metabolic, infectious and endocrine investigations were also negative.
One year later, a recurrent situation happened during a second pregnancy, ending with an early medical termination of pregnancy at 22 GA based on the presence of a hydramnios and a lack of fetal movement during ultrasound monitoring (Figure 2A). Exome sequencing was then performed for both siblings, and based on the familial recurrence, a recessive transmission was suspected. The analysis identified the TNNC2(NM_003279.3):c.314 + 1G > C p.(?) variant in both patients at the homozygous state. Sanger sequencing further confirmed the presence of this variant and inheritance from both heterozygous parents (Figure 2B).
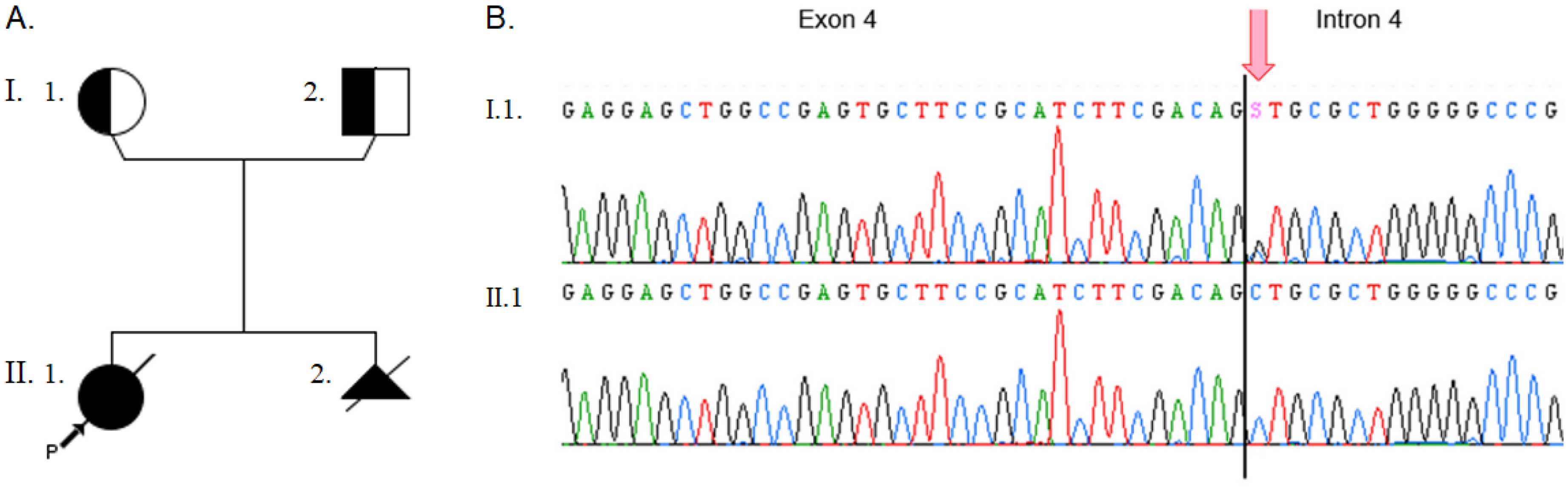
Phenotypes and genotypes. A. Patient pedigree. Half symbols indicate heterozygous status. Black symbols indicate homozygous status. Proband is indicated with the black arrow. B. Electropherograms from Sanger sequencing. The arrow indicates the c.314 + 1 position at the beginning of intron 4. Representative sequences are shown for one heterozygous parent (top) and one homozygous patient (bottom). S symbol represents the G/C substitution according to IUPAC nomenclature.
To this day, this variant has been reported with an allele count of 1 at the heterozygous state in population database (GnomAD v.4.1 3 ). This substitution is located on a consensual splice site and computational biology algorithms predicted a disruption of the donor site of exon 4 (MaxEnt mutscore 0.83 vs refscore 9.1 4 , SpliceAI donor loss score 0.99 5 ). RNAseq transcript study was then performed from the muscle RNA. It confirmed the absence of a normal splicing process due to the absence of a physiological splice junction, leading to an altered mRNA with different alternative splicing including the skipping of exon 4 and an intronic retention of 88 pb in intron 4 (Figure 3). Both could be responsible for a nonsense mediated mRNA decay (NMD), and the absence of wild type fast twitch skeletal muscle troponin protein.
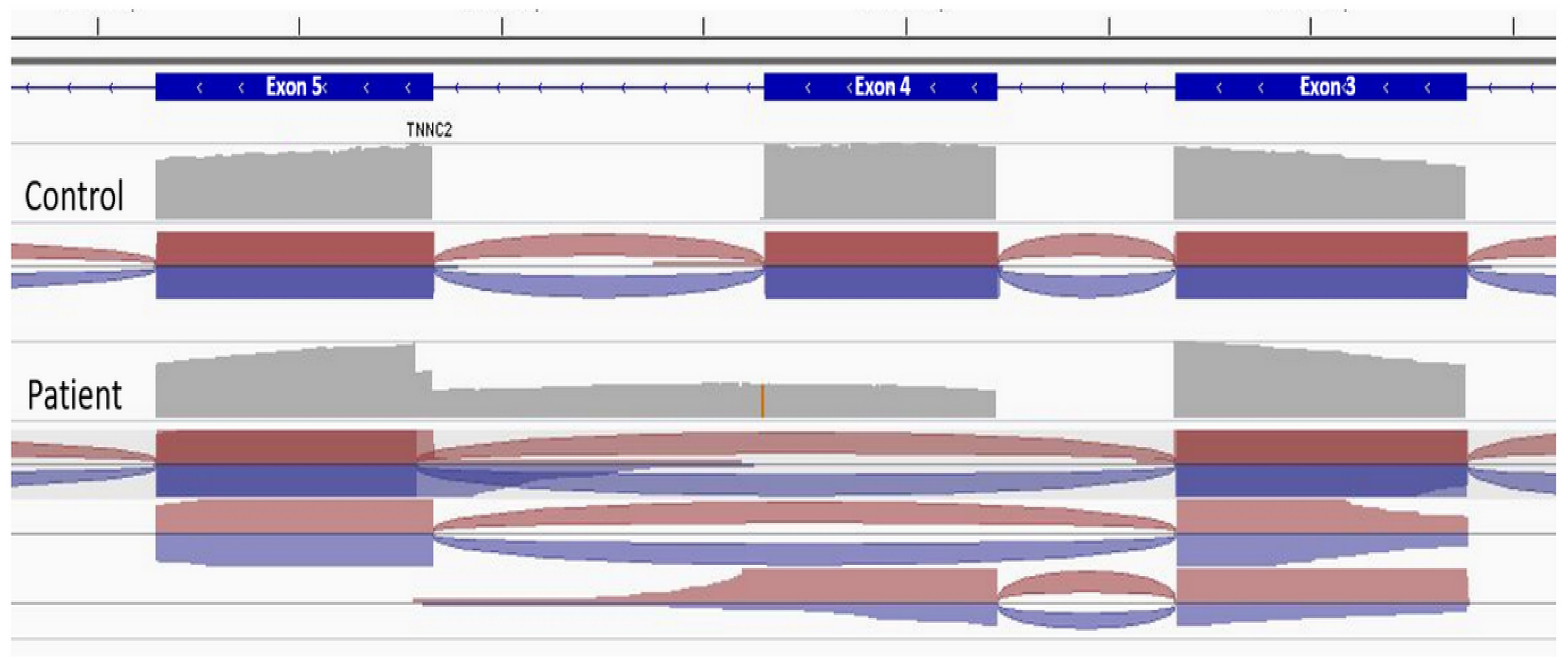
Splice IGV view of transcript analysis. RNAseq analyses were performed from the patient muscular transcript and from an unrelated control. Analysis from the control (top) shows physiological splicing of intronic sequences. The analysis from the patient's muscular transcript (bottom) shows two scenarii of abnormal splicing with a total exon 4 skipping (115 pb) or a retention of 88 pb corresponding to intron 4. Both should create a nonsense mediated mRNA decay or at least induce a shift in the reading frame leading to the absence of normal TNNC2 protein.
Discussion
The sarcomere contraction complex is a crucial component for fetal development, and many variants in muscle proteins have been shown to induce lethal forms of congenital myopathies. 6 For some of these myopathies, both dominant and recessive inheritance were reported. Congenital myopathies are defined by structural alteration and disorganization of muscle fibers and it is well established that biallelic loss-of-function variants can lead to such disorganization and defects in muscle contraction, as shown for genes like ACTA1, TPM3, TNNT1, TNNT3, MYH2 or MYH7.7,8 Hence, a similar mechanism for a recessive pathology should be considered for TNNC2. As transcript study showed an alteration of normal splicing, it is likely that the TNNC2(NM_003279.3):c.314 + 1G > C p.(?) variant should lead to a dysfunctional or absent protein that could explain the phenotype. So far, no lethal cases have been reported in dominant forms of myopathies due to TNNC2 but recently, TNNC2 has been included in a set of genes involved in major vital functions and associated with sudden death occurrence by machine learning, 9 suggesting that a recessive transmission of variant in the gene could be lethal. In addition, recent findings regarding TNNI1, a troponin-coding gene close to TNNC2, also identified both dominant gain-of-function variants and recessive loss-of-function variants in multiple families with various clinical muscular manifestations that have been attributed to the different inherent pathogenic mechanisms. 10
Of note, animal studies on zebrafish have shown that downregulation of TNNC2 using specific antisense morpholinos lead to an impairment in skeletal muscle function, decreased touch-response and an ability to swim for short distance only. 11 Importantly, no cardiac damage was observed when downregulating TNNC2 alone, whereas downregulation of its paralog TNNC1 led to heart and ventricle injuries. In humans, few pathogenic variants of TNNC2 are known but recently, Van de Locht et al. identified two heterozygous missense variants in two unrelated Caucasian families showing signs of muscle deficiency and motor difficulties. 12 Authors have linked these muscle deficiencies to an impaired sensitivity to calcium on myofibers with TNNC2 variants. The patients described in this study presented variable signs of facial and respiratory involvement. Interestingly, the onset was based on maternal polyhydramnios with severe hypotonia and breathing difficulties at birth. Muscle biopsies also showed hypertrophy of slow-twitch myofibers that could represent a functional compensation following the TNNC2 protein deficiency in fast-twitch fibers.
Altogether, these findings are consistent with the involvement of TNNC2 variants in severe neonatal forms of muscle deficiency. Although no other family with biallelic variant in the TNNC2 gene has been identified so far, it has already been reported that severe recessive forms of myopathies enlarge the spectrum of phenotypes associated with usually dominant transmission in a gene encoding a sarcomeric protein.13,14 Our report points to a recurrent phenotype in a family with no history of consanguinity, in which the segregation of a TNNC2 loss-of-function variant is compatible with a recessive form of myopathy associated with this gene considering the essential role of TNNC2 in muscle contraction. Nevertheless, further identification of biallelic pathogenic variants in TNNC2 are needed from additional cases to establish a recessive transmission. In addition, no complementary exploration was possible at the protein level, hence limiting the complete understanding of the mechanistic pathway underlying this case. We believe that this report should pave the way for additional evidences about this gene and its pathogenic pathways.
Conclusion
In conclusion, we report here the first case of TNNC2 homozygous loss-of-function variant leading to two pregnancies with unfavorable outcomes for the family. This case expands the spectrum of TNNC2 variants with a newly described recessive severe form of myopathy.
Material and methods
Immunohistochemestry and biopsy analysis
Open muscle biopsy was performed on quadriceps vastus medialis. Ten μm thick cryostat sections (Cryostar Nx70, Thermoscientific) were frozen and stained with haematoxylin and eosin (HE), modified Gomori trichrome (GT), reduced nicotinamide adenine dinucleotide dehydrogenase-tetrazolium reductase (NADH-TR) and ATPase pH 4.35 following standardized methods. They were analyzed with histochemical and histoenzymatic technics with light and electron microscopy (Leica ICC 50).
Molecular biology
Fetal DNA was extracted from amniotic liquid sample using trizol reagent (Invitrogen, Life Technologies SAS) and total RNA was extracted from muscle biospy with the Ambion PureLink kit (Invitrogen, Life Technologies SAS) according to the manufacturer's instructions. Genetic materials were quantified by absorbance measurement (Nanodrop, Thermofisher). Exome sequencing was performed by next-generation sequencing (NGS) on Illumina Seq500. Sanger sequencing was performed on ABI 3500XL with the following primers : 4F- ATCGAGGAGGTGGATGAGG and 4R- CCGTCTTTCATCAGAGATTCG. cDNAs of the TNNC2 transcripts were produced from mRNA extracted from muscle biopsies. The total transcripts were amplified, fragmented and sequenced on a S5 platform after library preparation (NEBNEXT New England Biolabs. 15 Transcript sequences and splice junctions were analyzed using NFCORE RNASEQ pipeline v.3.12 16 and IGV.
Footnotes
Aknowledgments
We thank all members of the family for their active participation in this case report. We also thank the CHUGA NGS platform for technical handling.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by the « Priority Research Programme on Rare Diseases » of the French Investments for the Future Programme.