
Abstract
Background:
Slow skeletal troponin T (ssTnT, TNNT1) is the tropomyosin-binding subunit of the troponin complex in the slow-twitch fibers of skeletal muscle. Exon 5 of TNNT1 is alternatively spliced, and retention of the 3’ region of intron 11 (exon 12’) has also been described. Variants in TNNT1 are known to cause nemaline myopathy (NM).
Objective:
To identify and further investigate the disease-causing variant in a patient with lethal NM.
Methods:
The genetic analyses included a gene panel, Sanger sequencing, whole-exome sequencing, and targeted array-CGH. Muscle biopsy was analyzed using routine histopathological methods. The alternative splicing of TNNT1 exon 12 in patient muscle was quantified from RNA sequencing data, and the protein expression was confirmed by western blot. Expression of ssTnT in patient muscle was studied by immunohistology.
Results:
The patient presented with arthrogryposis, stiffness, respiratory insufficiency, and minimal spontaneous movements. Histopathology showed hypotrophy and predominance of type II fibers, perimysial connective tissue accumulation, and nemaline bodies. The patient was homozygous for the TNNT1 missense variant (NM_003283.6:c.653C > G, p.(Pro218Arg), NM_ 001126132.3:c.612-7C > G), predicted to disrupt splicing. RNA-seq revealed inclusion of exon 12’ in 49.85% of transcripts, whereas in controls exon 12’ was not expressed. Exon 12’ expression on the protein level was confirmed by western blot. Immunohistology showed strong ssTnT expression in remaining type I fibers, and low expression in type IIA fibers.
Conclusions:
The c.653C > G variant was shown to alter TNNT1 splicing. The results suggest a novel pathogenetic mechanism involving abnormal expression of a troponin T isoform.
Introduction
Nemaline myopathies (NM) are a group of disorders characterized by generalized muscle weakness and Z-disc-derived protein aggregates, called nemaline bodies or rods, in skeletal muscle fibers. The clinical features of NM are heterogenous and the severity ranges from mild to lethal forms. 1 At least 12 causative genes have been identified to date. 1 Variants in TNNT1 (MIM: *191041) cause nemaline myopathy 5 (NEM5A, B, and C, MIM: #605355, #620386, #620389), originally identified among the Old Order Amish. 2 Since then, at least 30 TNNT1 variants have been published in association with the disease (Supplementary Figure 1).3–22 Most of the variants are loss-of-function variants inherited in homozygosity or compound heterozygosity, but two dominant variants have also been described.7,12,23
Characteristic features of severe infantile TNNT1-NM (NEM5A) are neonatal hypotonia, progressive muscle weakness, atrophy, and contractures. Affected children usually die of respiratory failure before the age of 2 years. 24 This severe phenotype is caused by deletions or truncating variants, whereas missense variants typically cause milder, non-lethal forms of the disease with later onset (NEM5B and NEM5C). The clinical characteristics of NEM5B and NEM5C are variable: some patients present only with mild weakness and remain ambulatory, while others require respiratory support or a wheelchair.7,10,12,16
TNNT1 encodes slow skeletal troponin T (ssTnT), which binds the troponin complex to tropomyosin in the slow twitch fibers of skeletal muscle. Cardiac muscle and fast twitch fibers of skeletal muscle express TNNT2 and TNNT3, respectively. 25 This restricted expression pattern forms only after birth; during embryonic and fetal development, skeletal muscle expresses mostly cardiac TNNT2 and an embryonic isoform of TNNT3, and the expression of TNNT1 in slow twitch fibers takes over during the first six months of life. 24 In addition to skeletal muscle, TNNT1 is also expressed in the fetal and adult heart. 26
Exon 5 of TNNT1 is alternatively spliced to create low and high molecular weight isoforms (LMW, NM_001126133.3, and HMW, NM_001126132.3, respectively). 27 In addition, a 48 bp inclusion of the 3’ region of intron 11 (exon 12’, NM_003283.6) has been reported.28,29 Overall, the functional role of these isoforms is unclear, but the HMW and LMW isoforms have been shown to differ in their troponin I (TnI) and tropomyosin binding affinities, as well as in their ability to create Ca2+ activated contractile force. 30 Some studies indicate that their expression pattern would be a physiological adaptation to exercise.29,31 No correlation with development has been established.27,31
Here, we identified a homozygous single nucleotide variant, NM_003283.6:c.653C > G, p.(Pro218Arg), NM_ 001126132.3:c.612-7C > G, on exon 12’ of TNNT1 in a patient with lethal NM. RNA sequencing (RNA-seq) from patient-muscle-derived total RNA revealed that exons 12 and 12’ were expressed in almost equal quantities, whereas in control muscles only exon 12 was expressed. In silico analyses indicated that the variant disrupts a splicing regulatory element, leading to the extension of exon 12.
Case report
Clinical findings
The patient was a girl born at 30 + 6 gestational weeks to consanguineous parents as the youngest child out of four in a family of African origin. The parents had experienced multiple miscarriages. Abnormal postures of the feet and hands and infrequent movements were noted antenatally. The birth weight was 1320 g (−1.4 SD) and head circumference 30.4 cm (+1.9 SD); the length could not be measured because of arthrogryposis with stiffness. In addition, she had hyperextended knees, unilateral hip luxation, and contractures in the 3rd and 4th fingers. Her spontaneous movements were minimal and she was unable to swallow. Cardiac and abdominal ultrasound were normal. Brain MRI showed minor bleeding in the cerebellar hemisphere but was otherwise normal. Slight asymmetry was shown at EEG, but there were no epileptic discharges. Ophthalmological examination gave normal results. Laboratory investigations showed low ALAT (8 U/l) concentrations, while CK was not quantified. The girl failed to establish spontaneous respiration and was dependent on mechanical ventilation, and was treated in the neonatal intensive care unit. Her respiratory drive showed no improvement, and she died at the age of one month.
Muscle histopathology
Muscle biopsy (quadriceps femoris) was performed at the age of one month. Histological examination (Figure 1A-D) showed perimysial accumulation of connective tissue. There was predominance of type II (fast) fibers, which were hypotrophic (diameter 6–10 μm). The size of the type I (slow) fibers was in the normal range, ca. 15 μm. Internal nuclei or necrosis were not present. Oxidative enzyme stains revealed no core structures or abnormal aggregations of organelles. Nemaline rods or cap structures were not observed at light microscopy. At electron microscopy (EM), however, numerous small, randomly distributed nemaline rods were observed within the myofibers (Figure 1E-F). Nonspecific myopathic changes with disorganized myofibrils, irregular and fragmented sarcomeres, streamed Z-lines, and mild glycogen accumulation, were also noted at EM (Figure 1G-H).
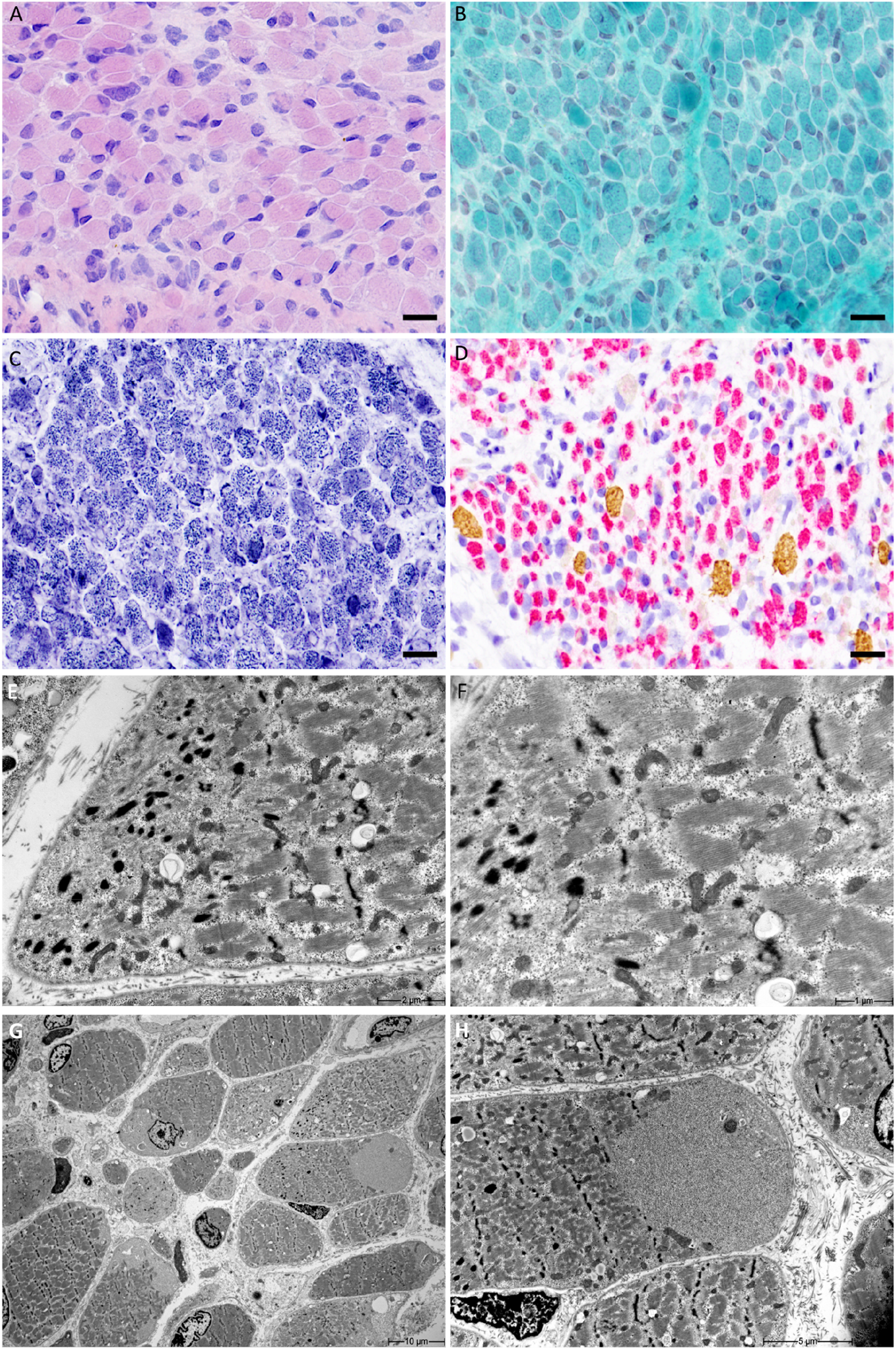
(A-D): Histological features of the muscle biopsy. A) Hematoxylin and eosin staining showed abnormal variation of myofiber size. B) Nemaline rods or mitochondrial accumulation were not present in Gömöri trichrome stains. C) The intermyofibrillar network was prominent, but core structures and organelle mislocalization were not displayed in NADH-TR stains. D) Immunohistochemical myosin heavy chain double staining showed predominance of hypotrophic type II fibers (brown (DAB) = type I slow fibers, red = type II fast fibers). Scale bar in A-D 20 μm. (E-H) Electron micrographs. E) Nemaline bodies were scattered within the myofibers and in the subsarcolemmal region. F) Higher magnification showing nemaline bodies at the I-band position, near the Z-line. G) Myofibers were disorganized, and there was slight glycogen accumulation. H) Sub-sacrolemmal glycogen accumulation in a muscle fiber, with nemaline bodies.
Genetic findings
The methods and the workflow are presented in Figure 2A. A homozygous single nucleotide variant in TNNT1 (NM_003283.6:c.653C > G, p.(Pro218Arg), NM_ 001126132.3:c.612-7C > G) was identified in routine diagnostics (Medical Genetics Center MGZ, Munich, Germany). There are three heterozygous carriers for this variant in the gnomAD v.4.1.0 database (https://gnomad.broadinstitute.org/, variant ID: 19-55134163-G-C in GRCh38). The variant is located on exon 12’ (Figure 2B). Most in silico prediction software predict the variant to be either benign or of uncertain significance (e.g., CADD 9.887, REVEL 0.02, AlphaMissense likely benign).
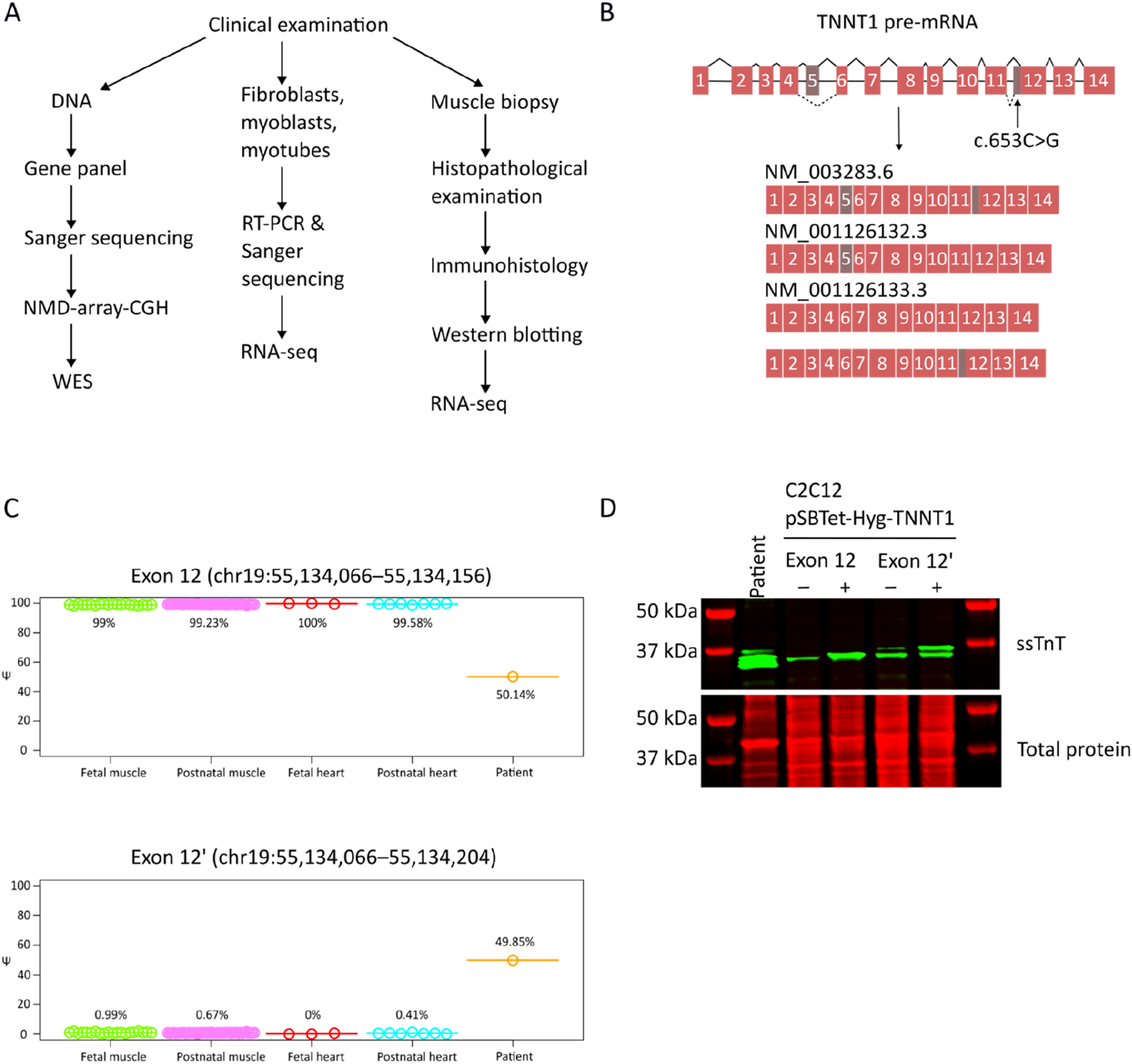
A) Outline of the methods used in this study. B) Schematic representation of the TNNT1 isoforms. Exon 5 is alternatively spliced to create high and low molecular weight isoforms. Intron 11 carries an alternative 3’ splice site. Out of the four potential isoforms, three have been annotated. C) Percentage Spliced In (PSI) values describing the proportion of the reads that support the alternative 3’ splice site upstream of exon 12 vs. the canonical 3’ splice site, across the patient and several (n = 45) fetal and postnatal cardiac and skeletal muscles. The PSI value in each sample is shown with a point and the average of the values across the sample groups are shown with horizontal lines. D) Western blot showing expression of TNNT1 in patient muscle. The C2C12-pSBTetHyg-TNNT1 cell lines serve as size markers for the isoforms. Expression of the constructs was not induced in the control samples (-).
The variant was confirmed in patient DNA using Sanger sequencing. Copy number analysis performed with a targeted array-CGH panel32,33 revealed no copy-number changes. No other candidate variants were found in whole-exome sequencing analysis (WES) (CeGaT GmbH, Tübingen, Germany). RT-PCR following Sanger sequencing was performed on patient fibroblast, myoblast and myotube cell lines as well as the muscle biopsy. The results indicated that TNNT1 exon 12’ was expressed, but the expression could not be quantified.
RNA-seq
Total RNA was extracted from the patient muscle biopsy with the RNeasy Fibrous Tissue Mini kit (Qiagen, Hilden, Germany) and sent for RNA-seq to Biomarker Technologies (BMK Gene, Münster, Germany). Illumina Stranded Total RNA Prep with rRNA depletion kit (Illumina, Inc., San Diego, CA, USA) was used for library preparation. The libraries were sequenced on the NovaSeq X platform (Illumina) (150 bp paired-end sequencing), producing about 44 million reads. The reads were mapped using STAR 2.7.7a 34 with the genome index generated from the Gencode.v39 human reference (GRCh38.p13, total number of genes: 61,533) using the STAR two-pass method. The alignment resulted in ∼90% uniquely mapped reads. Read summarization was performed using HTSeq. 35 The sequence fragments supporting the existence of each of the two 3’ splice sites of TNNT1 intron 11 were counted using IntEREst (V1.24.0) (https://bioconductor.org/packages/release/bioc/html/IntEREst.html). 36 The Percentage Spliced In (PSI) values associated with the alternative 3’ splice sites in the patient sample were also measured using IntEREst. These values were compared with similarly measured PSI values that were obtained by analysis of RNA-seq data from 45 postnatal muscles, 7 postnatal hearts, 20 fetal muscles, and 3 fetal hearts (Gene Expression Omnibus accession number: GSE270408). The splicing analysis revealed that exons 12 and 12’ were almost equally expressed (50.14% and 49.85%, respectively) in the patient muscle, whereas only exon 12 was expressed in the controls (Figure 2C).
To investigate the mechanism by which the variant affects splicing, the variant was analyzed using ESEfinder3.0 (https://esefinder.ahc.umn.edu/cgi-bin/tools/ESE3/esefinder.cgi),37,38 Human Splicing Finder 3.1 (HSF3.1) (http://www.umd.be/HSF3/), 39 and HExoSplice (http://bioinfo.univ-rouen.fr/HExoSplice/) 40 using their respective default settings. All three tools predicted that the variant disturbs exonic splicing enhancer/silencer (ESE/ESS) sites (Supplementary Figure 2A). ESEfinder3.0 returned five SR and two hnRNP sites for the wild-type sequence, and only two SR and no hnRNP sites for the variant. HSF3.1 predicted a significant alteration of the ESE/ESS motif ratio, with 11 broken ESE sites, one broken ESS site, one novel ESE site, and one novel ESS site. HExoSplice returned a positive ESRseq score, indicating a potential increase in exon inclusion.
C2C12-TNNT1 cell lines and western blot analysis
The HMW TNNT1 isoform with either exon 12 or exon 12’ with the c.653C > G variant was cloned to pSBTet-Hyg (a kind gift from Eric Kowarz; Addgene plasmid #60508; RRID: Addgene_60508). 41 To create C2C12 cells with inducible TNNT1 expression, C2C12 myoblasts were cotransfected with pSBTet-Hyg-TNNT1 and pCMV(CAT)T7-SB100 (a kind gift from Zsuzsanna Izsvák; Addgene plasmid #34879,RRID: Addgene_34879) 42 and selected with 200 µg/ml hygromycin-B. The cells were plated on ultra-compliant gelatin hydrogels (2.5% porcine skin gelatin (G2625, Sigma-Aldrich, Saint Louis, MO, USA) and ∼ 10 U/mL microbial transglutaminase (Bindly TI, BDF Natural Ingredients S.L., Spain) as described in 43 ), and differentiated into myotubes for 20 days under low-serum conditions (pyruvate-free DMEM with 2% heat-inactivated horse serum (Thermo Fisher Scientific, Waltham, MA, USA), 2 mM L-glutamine (Thermo Fisher Scientific), PS (100 units/mL penicillin, 100 μg/mL streptomycin), and 10% OPTI-MEM (Thermo Fisher Scientific)). The expression of the constructs was induced with 20 µg/ml doxycycline starting on differentiation day 3. The myotubes were lysed in RIPA buffer, combined with 2× Laemmli sample buffer and heated at 95 °C for 5 min. 10 µl of the lysate was used for western blot analysis.
The patient muscle biopsy was homogenized in sample buffer containing 0.5 M Tris-HCl pH 6.8, 4% SDS, 8% glycerol, 10% β-mercaptoethanol, and bromophenol blue. The homogenate was heated at 95 °C for 5 min and centrifuged at 13 000 rpm for 5 min. 6 µl of the supernatant was used for western blot analysis.
Proteins were separated in 4–15% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-Rad Laboratories, Hercules, CA, USA), and transferred on a nitrocellulose membrane with the Trans-Blot Turbo system (Bio-Rad) using the mixed MW program. Total protein was stained with Revert 700 Total Protein Stain (Li-Cor Biosciences, Lincoln, NE, USA). The membrane was incubated with rabbit polyclonal anti-TNNT1 antibody (HPA058448, Sigma-Aldrich; RRID: AB_2683720) at a 1:10 000 dilution at +4 °C overnight and with Alexa Fluor Plus 800 goat anti-rabbit IgG (Thermo Fisher Scientific ;RRID: AB_2633284) for 1 h at room temperature. The membrane was scanned with the Odyssey M Imaging System (Li-Cor).
The western blot analysis confirmed protein level expression of TNNT1 exon 12’ in the patient's muscle (Figure 2D).
Immunohistology
Immunofluorescence microscopy was used to assess the ssTnT expression pattern in the patient muscle biopsy at the protein level. 10 μm cryosections were fixed in 4% PFA for 10 min, permeabilized in 0.1% Triton X-100 for 20 min, and blocked in 10% Normal Goat Serum (50062Z, Life Technologies, Carlsbad, CA, USA) with 0.1% BSA for 1 h. Sections were incubated overnight at 4°C with mouse monoclonal anti-MYH I (slow, type I fibers) antibody (A4.951, sc-53090, Santa Cruz Biotechnology, Dallas, TX, USA; RRID: AB_2147279) diluted 1:25 or mouse monoclonal anti-MyH IIA (fast, type IIA fibers) antibody (SC71, DSHB, Iowa City, IA, USA; RRID: AB_2147165) diluted 1:25, each combined with rabbit polyclonal anti-TNNT1 antibody (HPA058448, Sigma-Aldrich; RRID: AB_2683720), diluted 1:500 or rabbit polyclonal anti-TNNT3 antibody (HPA037810, Sigma-Aldrich; RRID: AB_10966217), diluted 1:500 in 5% goat serum with 0.1% BSA and 0.1% Triton X-100. Alexa Fluor Goat anti-Mouse 647 (A21237, RRID: AB_1500743) was used as the secondary antibody for the MyHs and Alexa Fluor Donkey anti-Rabbit 488 (A11034, RRID: AB_2576217) for the TNNTs (Life Technologies, 1:500 each in 10% Normal Goat Serum).
Expression of ssTnT was strong in the few type I (slow) fibers present in the section (Figure 3A). Additionally, low expression was observed in the type IIA (fast) fibers, in which only the fast skeletal TnT (fsTnT) is normally expressed (Figure 3B, arrow). In the normal adult control (Figure 3C), ssTnT was expressed only in the type I fibers. The type IIA fibers expressed also fsTnT, as expected (Figure 3D). In contrast with the previously reported patients with severe NM (1-year-old disease control, Figure 3E), caused by the truncating TNNT1 variant p.(Glu180Ter)2,24,44,45 (Laitila et al. unpublished data), fsTnT was not expressed in the type I fibers. Compared with both the normal adult (Figure 3C, F) and the 1-year-old disease control (Figure 3E), all fibers, especially the type II fibers, were small. In the patient sample, however, the type I fibers were larger than the type IIA fibers.
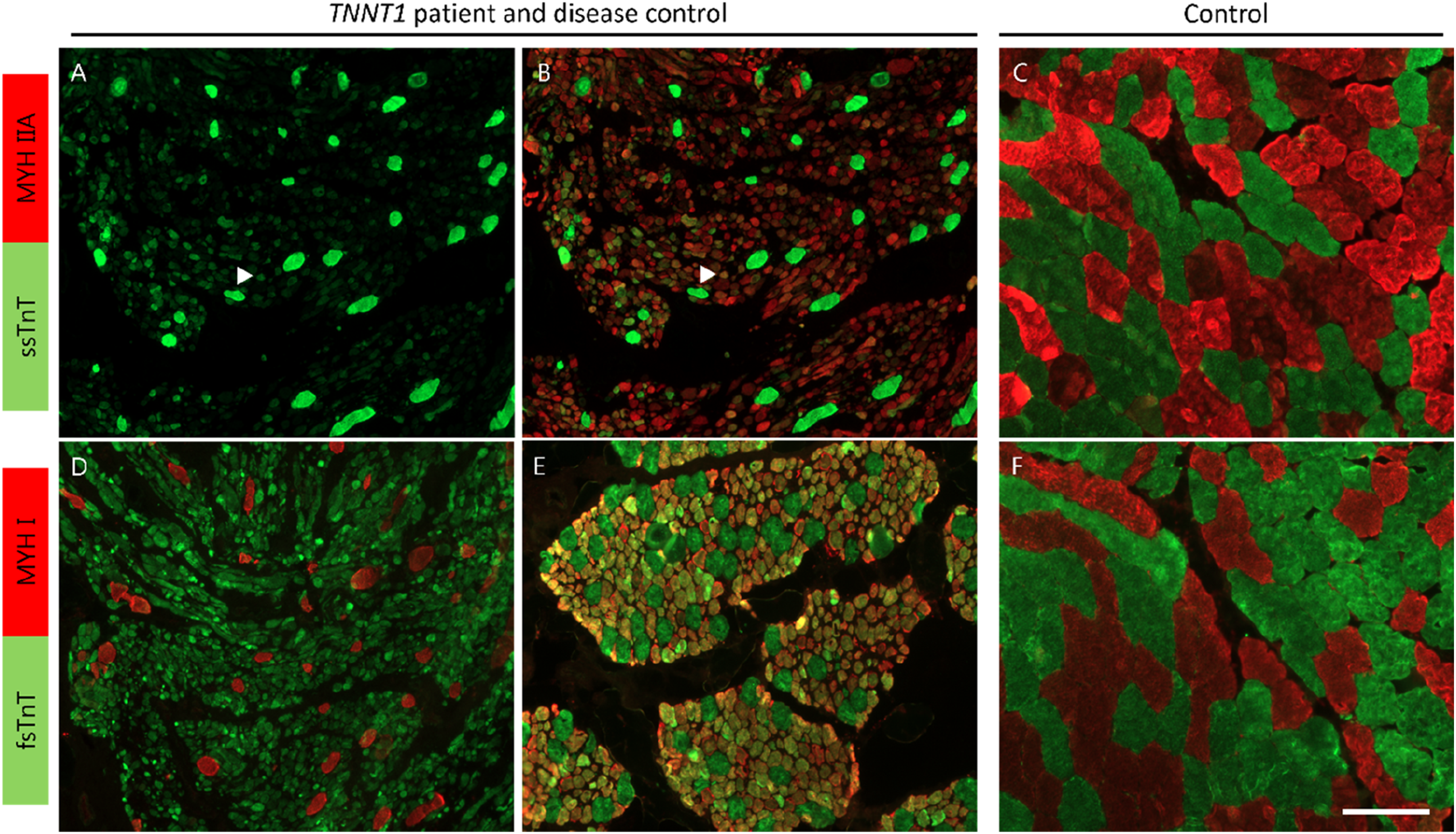
Slow skeletal TnT (ssTnT) and fast skeletal TnT (fsTnT) expression patterns in our patient muscle (A,B,D), a disease control (E), and normal control (C,F). A) The expression of ssTnT was strongest in the few MyH I (slow) fibers present in the biopsy. B) The ssTnT expression was also spread out to some Myh IIA (fast) fibers (arrow), although the expression was weaker than in the slow fibers. C) In the control biopsy, ssTnT was expressed in the slow fibers only. D) The type IIA fibers also expressed fsTnT, while no expression of the fast form was detected in the slow fibers. E) Compensatory expression of fsTnT in slow fibers in a patient with severe NM caused by the nonsense variant p.(Glu180Ter). In our patient, the expression intensity of fsTnT varied across the type IIA fibers, contrary to the type IIA fibers (green) in the patient control (E) and the normal control (F), where a more uniform expression pattern was seen. Scalebar 100 µm.
Mant-ATP chase expreriments
Control myofibers (vastus lateralis) were obtained from two individuals (29 year-old male and 35 year-old female). The sample preparation steps and Mant-ATP chase experiments were performed as previously described in Ranu & Laitila et al. (2022), 46 with a 10x objective on a Zeiss Axio Observer 3 fluorescence microscope with a Colibri 5 led detector, combined with a Zeiss Axiocam 705 mono camera, using Zen software (Zeiss, Oberkochen, Baden-Württemberg, Germany). While the percentage of myosin heads in the disordered-relaxed state (DRX) has been found to be elevated in other severe TNNT1-NM cases, 45 a balanced ratio was observed between the disordered-relaxed (DRX) and super-relaxed state (SRX = 100 – DRX%) in this patient (Supplementary Figure 2B). The time constant associated with DRX (T1) was decreased, however, (Supplementary Figure 2C), which suggests a faster ATP turnover rate in the patient myofibers compared with control myofibers.
Discussion
Here, we describe a patient with lethal congenital myopathy, who was found to have abnormal expression of TNNT1 exon 12’. The clinical picture of the patient was mostly consistent with TNNT1-NM, with minimal spontaneous movements, contractures, stiffness, and respiratory insufficiency. In contrast to most described TNNT1-NM patients, however, the disease onset in our patient occurred prenatally. Usually, the patients are born healthy, as the expression of TNNT1 replaces the immature forms of TNNT2 and TNNT3 in skeletal muscle only during the first six months of life. 24 In addition to our patient, only a few cases with prenatal onset have been described.5,18,19
The muscle biopsy of our patient showed several histopathological features unusual in TNNT1-NM. Immunohistochemistry for slow and fast myosin displayed predominance and hypotrophy of type II fibers. Typically, in TNNT1-NM, the type II fibers are hypertrophic, while the type I fibers are small and more abundant 24 (Laitila et al. unpublished data). Due to the very young age of the patient, however, interpretation of the fiber sizes might be skewed by Wohlfahrt B fibers, which are large type I fibers produced in primary myogenesis, and likely constitute at least a part of the type I fibers in the patient. Therefore, it is possible that the type II fibers only appear hypotrophic, when in fact all fibers produced in secondary myogenesis are uniformly small. No nemaline bodies were observed in the muscle biopsy of our patient at light microscopy, but numerous small nemaline bodies were seen in EM. The nemaline bodies were too small to be detected at light microscopy (the average width of the nemaline bodies being 170 nm).
Immunohistological staining showed that the expression of ssTnT was strong in the remaining type I fibers (Figure 3A), and interestingly, low levels were detected also in type IIA fibers, normally only expressing fsTnT (Figure 3B). No compensatory expression of fsTnT in the slow fibers was observed, contrary to previously reported patients, likely contributing to the severity of the disease (Figure 3D, E).
Western blotting confirmed the expression of the exon 12’ -encoded peptide segment at the protein level (Figure 2D). In the previously described TNNT1-NM cases, the severity of the clinical picture correlates with residual ssTnT protein levels, with the most severe phenotypes resulting from total absence of ssTnT. Our results indicate a novel pathogenetic mechanism, as the phenotype was lethal despite strong residual expression. This is further supported by the balanced ratio of SRX/DRX of relaxed myosin observed in the patient myofibers (Supplementary Figure 2B), compared with the elevated DRX percentage that has been seen in other severe TNNT1-NM cases lacking ssTnT expression (Laitila et al., unpublished data). Ranu & Laitila et al. (2022) 46 showed that the DRX percentage was elevated in NEB-NM and ACTA1-NM, but not in TPM2/3-NM, indicating variability between NM patients harboring mutations in different genes. More experiments analyzing the distribution of the conformational states of relaxed myosin in a broader range of NM patients are needed, however, to gain more insight into the pathogenetic mechanisms related to relaxed energy expenditure.
The major TNNT1 isoform in adult muscle is the HMW isoform, whereas the expression of the LMW isoform increases in response to certain pathophysiological conditions, such as Charcot-Marie-Tooth type 1 and dominant TNNT1 myopathy, and in response to resistance training.7,29–31 The expression of exon 12’ was reported by Gahlmann et al. (1987), 28 but very few studies have since described this event. Samson et al. (1994) cloned adult human quadriceps cDNA, which did not include the exon 12 extension. 47 Likewise, Konersman et al. (2017) 7 or Pellerin et al. (2020) 16 did not observe exon 12’ in their patients or controls. Exon 12’ was detected at a very low level, however, in two patients with dominant TNNT1 myopathy and in healthy controls, 12 but significant quantities have not been detected reliably. 29 Correspondingly, the exon 12 extension was not seen in our control cohorts (Figure 2C).
We hypothesize that the lethal congenital myopathy in this patient was caused by the abnormal expression of TNNT1 exon 12’ due to the c.653C > G variant. According to the in silico splice site analyses with ESEfinder3.0, HSF3.1, and HExoSplice, the variant is located in a regulatory region which contains ESE and ESS sites (Supplementary Figure 2A). These sites are important auxiliary sequences that bind SR and hnRNP proteins. The binding of SR proteins to ESE sequences promotes splicing, whereas hnRNPs recognize ESS sites and inhibit splicing. 39 The loss of SR and hnRNP binding sites, as predicted by the three software that we used, likely leads to the use of an alternative 3’ splice site, extending exon 12 by 48 bp. This splicing abnormality seems to occur at a low level in normal muscle,12,28 suggesting a threshold above which the expression is no longer tolerated. It is probable that the patient's healthy parents, as carriers of the c.653C > G variant, express the exon 12’ at some level; however, they were not available for analysis.
Troponin T plays an essential role in sarcomere assembly and maintenance. Knockdown of troponin T genes in zebrafish embryos resulted in the loss of organized sarcomere structure, while partial disruption of troponin T lead to the disintegration of initially formed myofibrils. 48 Similarly, in Drosophila melanogaster, troponin T mutations that reduced the levels of the protein disrupted myofibrillar structure, so that no sarcomeres were formed. 49 Like reduced expression, overexpression of troponin T leads to impaired sarcomere assembly. In D. melanogaster, overexpression of troponin T provoked a decrease in other thin filament mRNA and protein levels, subsequently disrupting myofibrillar assembly. 50 Excessively high expression of the TNNT1 isoform with exon 12’ in our patient may interfere with thin filament assembly or function. The 16 extra amino acids (amino acids 205–220 in NP_003274.3) are located in the IT arm, which contains the troponin I binding site. 23 Furthermore, the tropomyosin binding site T2 is located near, at amino acids 179–203. 51 It is possible that the binding of troponin T to troponin I and tropomyosin is disrupted by the addition of these residues.
In conclusion, we report a case of lethal nemaline myopathy caused by an imbalance of ssTnT isoforms, due to a homozygous single nucleotide variant disrupting a splicing regulatory element. Our results expand the spectrum of pathogenic TNNT1 variants.
The study has been approved by the Ethics Committee of the Children's Hospital, and the Helsinki University Hospital, University of Helsinki, Helsinki, Finland (9/2021).
Supplemental Material
sj-docx-1-jnd-10.1177_22143602251339569 - Supplemental material for A homozygous single-nucleotide variant in TNNT1 causes abnormal troponin T isoform expression in a patient with severe nemaline myopathy: A case report
Supplemental material, sj-docx-1-jnd-10.1177_22143602251339569 for A homozygous single-nucleotide variant in TNNT1 causes abnormal troponin T isoform expression in a patient with severe nemaline myopathy: A case report by Milla Laarne, Ali Oghabian, Jenni Laitila, Pirjo Isohanni, Olli Tynninen, Fang Zhao, Fanny Rostedt, Jaakko Sarparanta, Lydia Sagath, Michael W Lawlor, Carina Wallgren-Pettersson, Vilma-Lotta Lehtokari and Katarina Pelin in Journal of Neuromuscular Diseases
Footnotes
Acknowledgements
We thank Marilotta Turunen and Helena Luque for technical assistance, Swethaa Natraj Gayathri for assistance with the RNA-seq samples and data analysis, and Dr Anders Paetau for expert histopathological analysis.
Funding
The research is funded by the Jane and Aatos Erkko Foundation, the Folkhälsan Research Foundation, Finska Läkaresällskapet, and Medicinska Understödsföreningen Liv och Hälsa. A.O. is funded by the Magnus Ehrnrooth foundation.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.